
In Parts 1 and 2 of our series, we introduced the rare disease (RD) ecosystem as a uniquely interrelated set of stakeholders and discussed the key challenges and considerations for successful RD clinical trials. We focused on (pre)registrational clinical trial design, but of course regulatory approval does not mark the end of evidence generation. Here in Part 3, we expand the discussion to cover continuous evidence generation for the rare disease therapeutic.
Ongoing evidence generation is particularly important for RD given the unique characteristics of therapeutic development and commercialization in this ecosystem. Often, the typically small registrational trials are not long enough or sufficiently statistically powered to address all these considerations, so continuous evidence generation trials must be appropriately designed to do so.

As in all diseases, lifecycle planning and ongoing evidence generation for RD should be oriented towards one of more strategic objectives that maximize the value of the therapeutic as well as the portfolio itself, e.g., the ACE framework.
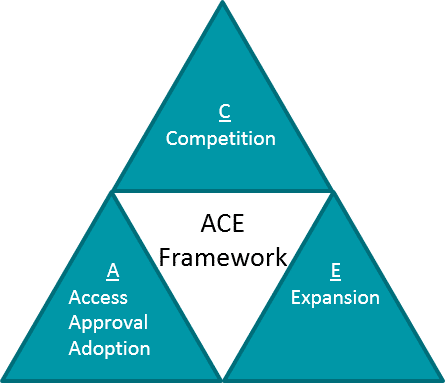
However, in RD there are unique considerations for each aspect of this framework:
- Approval, Access, Adoption: In RD, additional supportive trials that can continue after approval must fill in any evidence gaps left by a potentially small and rapid registrational trial.
- Competition: In some RD, there are multiple competitors either in the market or in development, so competition exists in ways similar to other disease areas. However, in other RD, there is no or limited standard of care, so the main “competition” is lack of awareness and / or alignment on differential diagnoses, diagnostic tools, and disease management. Continuous evidence generation trials may therefore need to generate foundational market education and awareness.
- Expansion: In RD, there is a spectrum where some therapeutics are precisely tailored and therefore limited to their lead indication, whereas others may expand into additional diseases. However, even the limited therapeutics may support platform or portfolio expansion.
A: Approval, Access, Adoption
Like other high unmet need disease areas, rare diseases frequently obtain an accelerated regulatory review and decision. When the decision is favorable, it may still be conditional – requiring further efficacy and / or safety data. In parallel, even with regulatory approval, payers may resist coverage based on the initial data package (particularly where there are concerns about cost-effectiveness relative to a high price), requiring further evidence generation. Therefore, initial follow-on studies are typically aimed at label confirmation and / or reimbursement negotiations.
These studies should be designed to address common expressed or anticipated concerns about the initial registrational dataset, for example:
- Further characterization of therapeutic impact on natural course of disease, to define efficacy and durability profile (especially important in RD with slow progression, or where part of the therapeutic value proposition is single or limited dosing)
- Confirming predictive power of surrogate endpoints (e.g., biomarkers) for clinical benefit
- Delivering larger n-values / statistical power to reinforce initial dataset
- Extending safety monitoring (especially important where therapeutics are expected to be used indefinitely to manage chronic conditions)
- Calculating HEOR impact (especially important for high-priced therapies)
Approval: Because the available patient population for trials is often quite limited in RD, these studies are often designed as extensions of the registrational study. As such, they need to be designed and planned at the same time as the registrational study. Important considerations here include pre-defining milestones and data thresholds under which to file “early” vs continue gathering data, as well as designing studies in such a way that the limited patient set is maximized (e.g., crossovers so all patients can receive therapy and contribute to efficacy / safety data). As with all accelerated approval attempts, there is also the risk that an initial approval fails to be confirmed, resulting in a label retraction.
Access: For those studies which are also intended to support payer negotiations, they may also include additional endpoints or outcomes measures which are relevant for HEOR. Examples may include the frequency of acute disease events requiring hospitalization (e.g., seizures), or the percentage of participants avoiding a disease progression milestone which requires a significant increase in care (like ventilator reliance or tube feeding), etc. Because many RD are progressive, it is particularly important for the RD company to have robust data to characterize the typical natural history of the disease. This is because in some cases the benefit of the therapeutic will not be that it improves symptoms, but rather that it slows progression.
Adoption: Once approval and access are in place, ongoing evidence generation becomes more focused on adoption, via deepening and broadening of the overall data package to support incorporation into guidelines and actual clinical practice. As ongoing evidence generation trials are started up in new sites, these activities also support local adoption as practitioners become more familiar with the disease and therapeutic, in addition to supporting the development of new Centers of Excellence and Key Opinion Leaders. The latter is particularly important if the RD company has other related pipeline products in development, as it provides a base from which to launch future clinical trials more efficiently.
C: Competition
Outside of RD, most therapeutics enter markets with established treatments, so the concept of competition is focused on differentiation from those. However, in most rare diseases, the new therapeutic is not entering a competitive space (because there isn’t any existing approved therapy). Instead, the main challenge is lack of knowledge about the disease itself. So, in RD, the concept of competition can also include market education and shaping (i.e., competing against lack of knowledge).
Ongoing evidence generation will be critical here to provide clarity not just on the efficacy / safety profile of the therapeutic, but the most impactful ways to diagnose, characterize or stratify the disease state, and track outcomes over time. This can be as foundational as establishing diagnostic and staging strategies where there is no current consensus, as well as broad educational efforts to identify these rare patients earlier in their treatment journey. As discussed above, this is also quite critical where a therapeutic works by slowing progression vs reversing the course of disease, as that declaration must be supported by a deep understanding of the natural history of untreated disease.
RD companies that are first to enter the market get to define the patient population and endpoints / outcomes measures, which can be beneficial to them where competitive follow-ons aim for a different niche that first must be proven. However, it can also enable competition to fast-follow if their therapeutic is suitable for the same population / endpoints.
In the subset of RD where inter-therapeutic competition is a factor (for example Cystic Fibrosis, Spinal Muscular Atrophy, Duchenne’s Muscular Dystrophy, Hemophilia), then the role of ongoing evidence generation in competitive positioning is similar to other therapeutic areas, namely differentiating on indication, efficacy, safety, and / or dosing, while still maintaining a beneficial pricing profile. RD companies operating in a competitive space should ensure that continuous evidence generation trials answer one or more of the following:
- Can they demonstrate an improved efficacy / safety / durability profile vs competition? E.g., for those RD that are chronic or in high risk populations, does the safety or durability enable differentiation
- Can they improve their dosing / administration profile vs competition? E.g., fewer doses, less invasive route of administration (oral vs injection)
- Can they relieve personal / caregiver / health system overall burden as compared to competition? This may include patient-reported outcomes associated with quality of life, and is especially important for chronic and progressive RD.
- Can they bolster market position by treating a broader proportion of patients than competition (without diluting their efficacy / safety profile)? Typical expansions involve including additional genotypes or disease subtypes, or accepting either older or younger patients. For newborns or infants, the safety profile becomes particularly important.
One illustrative example is Spinal Muscular Atrophy (SMA), where three recently-approved therapeutics are using a variety of continuous evidence generation trials to compete.
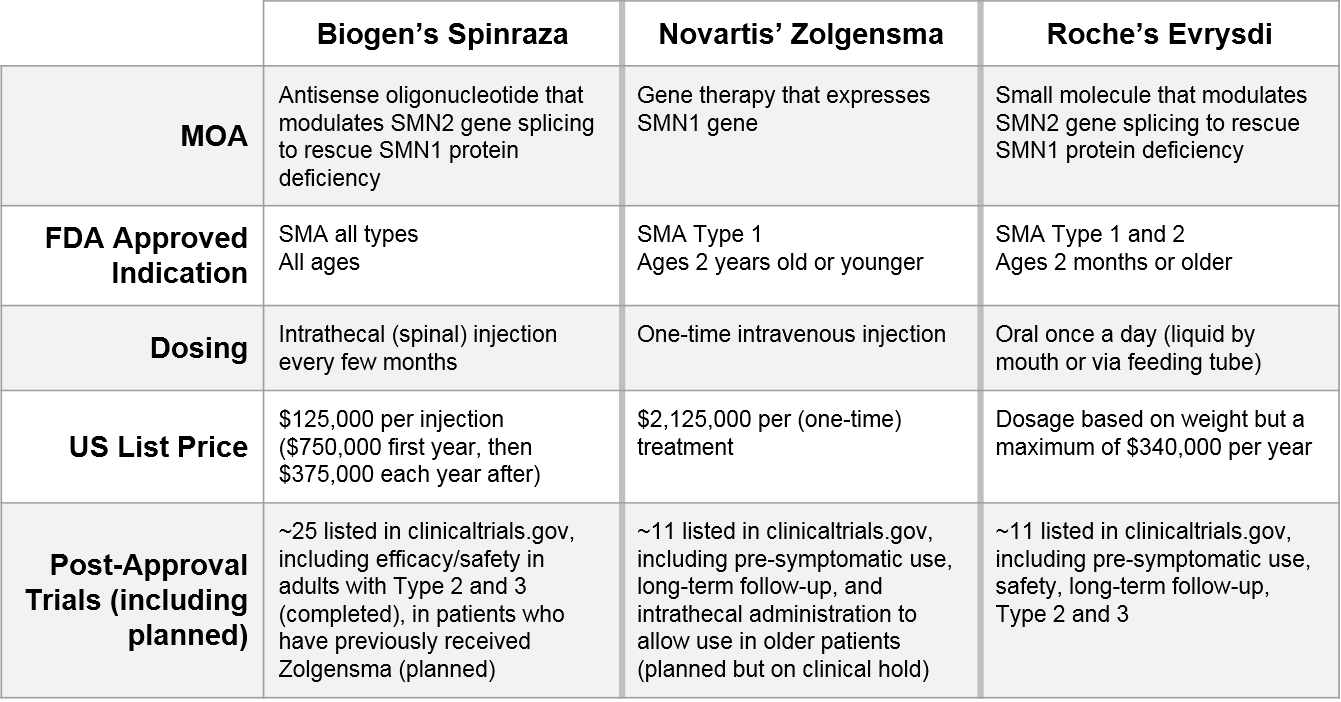
Some of these factors (the MOA and the initial indication and dosing administration) are fixed and cannot be changed, but you can see from the portfolio of ongoing trials how each company is seeking to differentiate and compete along different dimensions:
- Indication:
- Biogen’s Spinraza has the broadest label to start with, but is also exploring use after Novartis’ Zolgensma (a sequenced regimen play)
- Novartis’ Zolgensma is exploring use in pre-symptomatic and older patients
- Roche’s Evrysdi is exploring use in pre-symptomatic and Type 3 patients, as well as in patients who have already received Spinraza or Zolgensma
- Efficacy / Durability / Safety:
- Novartis’ Zolgensma and Roche’s Evrysdi are both pursuing longer-term efficacy and safety data (Biogen’s Spinraza has already completed this follow-up)
- Dosing / Route of Administration:
- Novartis’ Zolgensma is pursuing an intrathecal administration (vs their current IV formulation) which would enable use in older patients
These existing and emerging post-registrational data packages, along with adjustments in pricing strategy and associated calculations of HEOR benefit and relief of caregiver / health system burden, will be key factors in how well each of these therapies is able to carve out and maintain a niche over time.
E: Expansion
As an RD company launches and positions a new therapeutic within its lead indication, in parallel the focus broadens to include expansion of the therapeutic as well the platform and portfolio (as relevant). Expansion can be considered along various dimensions:
Within lead disease space: The most straightforward version of this type of expansion is geographic, where clinical trials are designed similarly to the initial approval trial (and often are running in parallel) but in various geographies. The next layer of this is label broadening within the initial disease space. This broadening can include additional ages (whether older or younger), additional genotypes or other measures of disease characterization, and / or use in the context of competition (for example in combination or sequence with other treatments). Here, it is important to take into account whether broadening the patient population will reduce the overall benefit / risk ratio which in turn can negatively impact the value proposition.
Into other diseases: If the therapeutic has a target / MOA which is relevant in additional diseases, those development programs can be initiated to follow the lead indication, taking advantage of already established endpoints and dosing / safety data. Speed is critical here, to take advantage of patent protections put in place at the time of first approval.
Repurposing of platform: For a platform-based therapeutic, e.g., a gene therapy, the initial approval generates a “backbone” where additional targets / MOAs can be swapped in, and then skip ahead through the initial characterization of efficacy and safety for the platform (which is independent from that of the therapeutic itself). This can enable acceleration of different variations into new disease spaces.
Boosting of portfolio: For an RD company that has multiple therapeutics in development within the same general space, there may be opportunities to develop and then leverage novel endpoints / outcomes measurements across related diseases. This type of evidence generation can then streamline development for subsequent therapeutics, as regulatory bodies, payers, and other key stakeholders now have experience in interpreting these data.
Key Stakeholder Engagement:
As with design of registrational clinical trials, key stakeholder engagement is critical for post-registrational ongoing evidence generation.
- Regulatory bodies: critical to engage early (at registrational trial stage) to understand which longer-term or additional endpoints may be required for confirmatory studies, and what data package will be sufficient for regulatory decision-making. In some new (to them) RD, they must also be supported in disease education to understand the natural history of the disease and outcomes in order to contextualize the benefit of the new therapeutic
- Payers: in addition to ensuring comfort with data quality (similar to regulatory bodies), there is the added dimension of providing evidence of value in the context of price – therefore it is important to engage and collaborate as HEOR and other value calculations are designed. As above, Payers may also require education in RD where no treatments have previously been available.
- Scientific Societies / Academia / Centers of Excellence (COE) / Key Opinion Leaders (KOLs): these players are critical in translating an initial approval into a new standard of care, and their perspective on registrational trial data gaps should inform ongoing evidence generation trial design to fill those gaps. This ongoing work also supports the development of new COEs and KOLs, as new centers and practitioners are brought on board via clinical trial participation.
- Patients / Caregivers / Patient Advocacy Groups: similar to their role in registrational clinical trial design, these stakeholders must also be involved in post-registrational trial design to ensure that the trial is designed appropriately to demonstrate clinical benefit (e.g., relevant endpoints, appropriate inclusion criteria) and is attractive to participate in (not unduly burdensome, guarantee or good odds of receiving the therapeutic, etc)
Conclusion
Rare disease evidence generation can be challenging in similar ways to registrational trials (small patient population, high need for stakeholder engagement and buy-in). However, it also offers enormous potential for ongoing market education and growth not just within the lead indication but sometimes into additional disease areas or across related therapeutics within a portfolio. In this paper and the previous we described key considerations for trial design. In our next paper, we will focus on the practical and logistical elements of trial implementation.








