
Introduction
Spinal Muscular Atrophy (SMA) is a devastating disease, but recent therapeutic advances are delivering major benefits for patients, as well as driving competition in a complex market landscape. In Part 1 of our series, we reviewed the disease biology and therapeutic approaches currently marketed to treat SMA. Here, we look to the future and discuss ongoing and potential advancements that will impact the SMA therapeutic and commercial landscape.
Despite the major advances of the last few years, we recognize remaining unmet needs for SMA patients not currently served by the existing therapeutics. Addressing these needs will require some combination of broader use of existing therapies as well as development of even more effective regimens. We anticipate several key trends that will address these unmet needs but also collectively increase market complexity for SMA. This will have implications for players in this space as well as any biopharma company working in a similarly complex disease area.
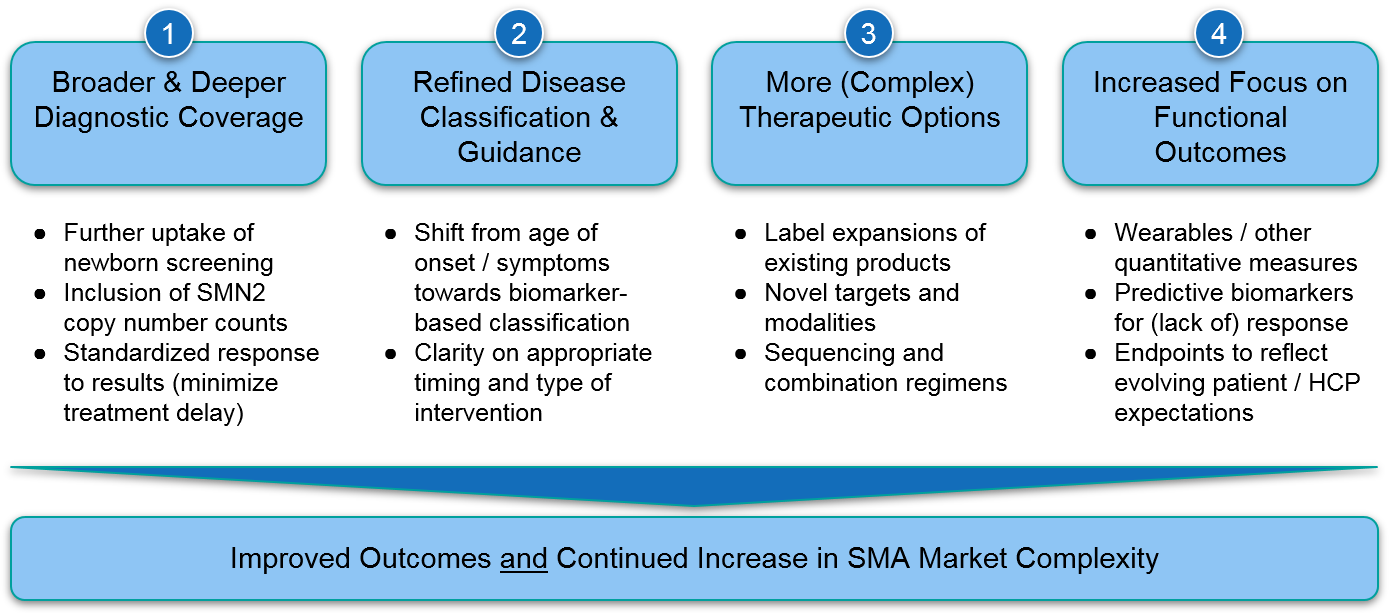
Trend 1) Broader & Deeper Diagnostic Coverage
Once motor neurons die and muscles atrophy due to loss of Survival Motor Neuron (SMN) protein, they cannot easily regain lost function. Therefore, it is critical to identify SMA as early as possible and intervene with an SMN protein boosting therapy that protects the remaining motor neurons. Often, a significant percentage of motor neurons have already died by the time symptoms are apparent, so early detection of the underlying genetic alterations (followed by swift therapeutic intervention) will ensure the best possible outcome for each individual.
For more details on the underlying biology of SMA and why genetic screening is critical, see Part 1 of our series. To summarize, motor neuron death in SMA is caused by SMN1 gene mutations, but is partially compensated for by SMN2 gene expression. SMN2 may exist as two or more copies in the genome, and the number of copies is inversely proportional to the severity of the disease. For example, infants with only 2 copies of SMN2 are either born symptomatic, or rapidly decline within weeks of birth, whereas individuals with 4 or more copies may only develop mild symptoms, if any. Therefore, it is important to know not just SMN1 mutational status, but how many copies of SMN2 are present.
At the moment, SMN1 mutation screening is part of the Recommended Uniform Screening Panel (RUSP), but only about ⅔ of US infants receive SMN1 testing. This contrasts with screening for the metabolic disorder PKU, which is government-mandated and therefore done for all infants at birth. Even for those newborns who do receive SMN1 screening, there is significant variation across institutions on how the results are incorporated into practice. For example, in some states the results are only sent to the pediatrician, who may be unfamiliar with SMA and not aware that time is of the essence for treatment to begin. Because most screening panels do not also include SMN2 copy number, there is also the risk of treatment delays for infants with only 2 copies of SMN2, who may suffer clinical decline within days or weeks.
As such, current genetic screens are sub-optimal in identifying SMA patients for timely intervention. However, because the connection between underlying genetics and disease progression is so clear, and because we have these existing effective interventions, we expect continued uptake of broader and more sensitive diagnostic screening tests which will in turn improve patient outcomes. However, biopharma may need to help accelerate these trends in order to identify rare SMA patients within the relatively narrow therapeutic window.
Implications for Biopharma: For players seeking to enter this or similar markets, it may be necessary to support broader access and / or development of better diagnostics to identify as many of these rare patients as possible. This may include
- Investing in clinician education regarding SMA
- Standardizing genetic screening protocols and methods of results interpretation to decrease the currently problematic variability in the patient journey
Additionally, financial support would increase patient access to testing until tests are approved, accessible, and reimbursed. Biopharma may also consider partnering with patient advocacy groups to bolster education as well as explore government-mandated inclusion of SMA testing into newborn screening.
Trend 2) Refined Disease Classification & Guidance
Given the increase in earlier diagnosis and the availability of effective therapeutic interventions, it is likely that over the next few years, disease classification will shift from the current model (based on age of onset and maximum function reached if untreated) to one based on the underlying genetics (i.e., SMN2 copy number) and the age / outcomes of treatment initiation.
Figure: Possible Evolution of SMA Disease Classification
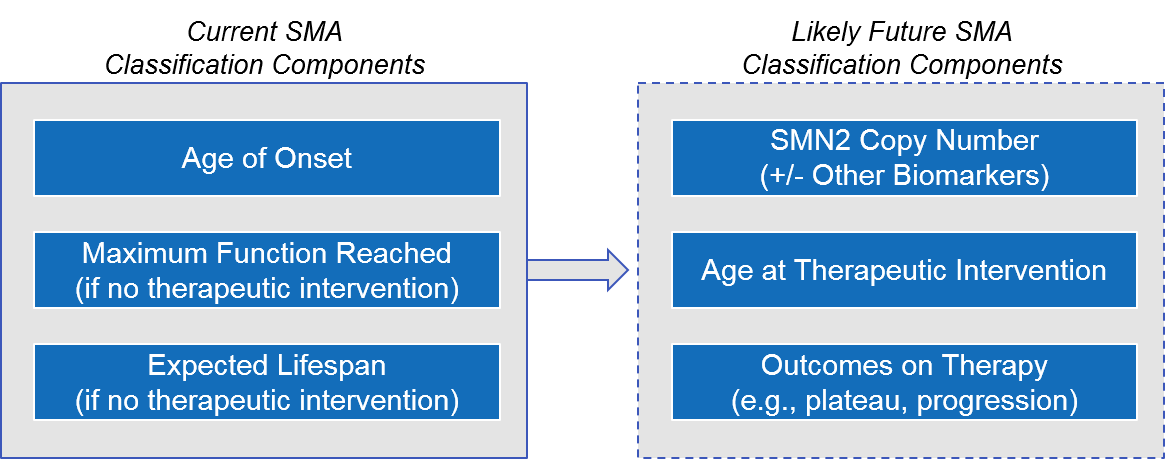
This evolving understanding of the underlying biology and real-world implications of highly effective treatments will have significant implications for treatment decision-makers, particularly while clinical data and labels catch up. For example, the FDA and EU labels for the approved therapeutics are restricted based on age and / or underlying genetics (the presence of SMN1 mutation and sometimes the copy number of SMN2).
Given the heterogeneity of the disease and its presentation, this means that some individuals are caught in a situation where none of the approved therapeutics is approved for them, based on age and / or underlying genetics. This is particularly true in Europe, as the EU label for both Zolgensma and Evrysdi are restricted by age, SMN1 mutation, and SMN2 copy number, as opposed to the US which is focused on age and / or SMN1 mutation. While the research and development strategies described above will hopefully expand the approved options, in the meantime clinicians must make some decisions in the absence of guidelines or robust clinical data. For example, given label language about infants being “at term”, there is some debate in the field about when to treat premature infants with genetically diagnosed SMA.
There is also uncertainty about treatment algorithms for both extremes of disease presentation. The most severe form of SMA (called Type 0 or prenatal) often kills in utero, and those infants born alive usually require ventilation at birth and have limited to no motor function. The approved therapeutics have not been well-tested in this setting, and it is unclear whether they will provide significant benefit to this population as the motor neurons have already been severely impacted by the time of treatment.
On the other end of the spectrum, SMA patients who are early in their disease but known to have multiple extra copies of the SMN2 gene (which helps compensate and reduces the disease severity) may not necessarily need therapeutic intervention, and in fact the benefit/risk ratio may not be favorable to them given side effects of treatment. However, if the therapeutic window is missed, then it is too late. So, in the absence of data and guidelines indicating that treatment can safely be skipped, clinicians will be motivated to treat these Type 4 patients.
For both populations – the least severe Type 4 and most severe Type 0, there is also the issue of insurance coverage and reimbursement given label language, as well as limited available data for providers to make their case.
Finally, we may consider an overarching category which includes the patients described above, but also those with non-SMN-driven disease (i.e., SMA with other underlying mutations) and those with SMN-driven disease who for whatever reason had limited response to therapy or have missed the therapeutic window for efficacy and now have more advanced and intractable disease. While we expect the percentage of patients in this group of “left behind” to shrink over time, the ones remaining will continue to need therapeutic support.
Implications for Biopharma: Clinical development programs will need to account for both the current and anticipated future disease classifications as they define their target patient population. For example, they may wish to stratify by SMN2 copy number (or other biomarkers) and be prepared to analyze outcomes based on timing of interventions. To develop compelling datasets that bridge this transition, biopharma can collaborate with expert clinicians to retrospectively interrogate existing data, as well as prospectively design trials where needed for label expansions or large shifts in clinical practice. Clinical development programs should also consider investing in novel therapies / regimens to expand into the remaining unmet need categories described above.
Trend 3) More (and More Complex) Therapy Options:
As in all active therapeutic areas, in SMA we see three overlapping arenas of therapeutic development involving expansion of existing therapies, entry of new therapies, and regimens involving various permutations of the two.
Figure: SMA Therapy Evolution and Illustrative Examples
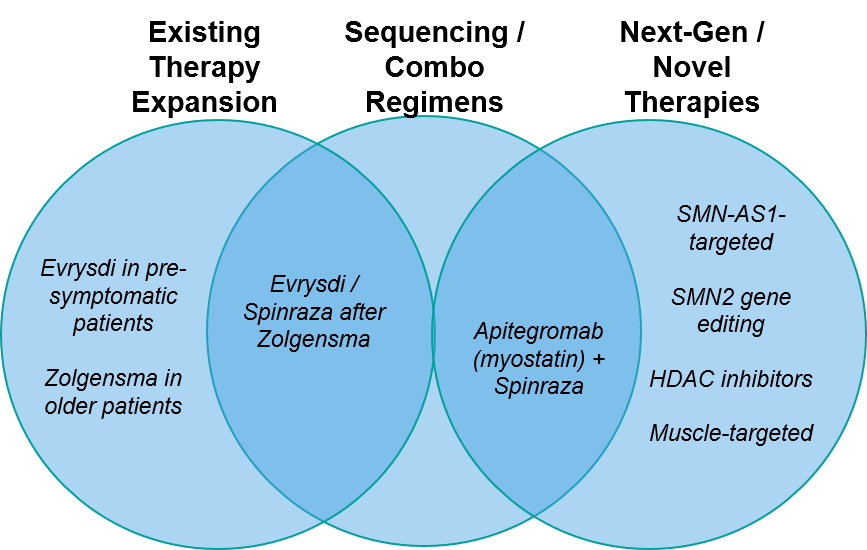
Existing Therapy Expansion:
Naturally, all three of the approved SMA therapies are actively exploring label expansions:
- Biogen’s antisense oligonucleotide splicing modifier Spinraza
- Novartis’ gene therapy Zolgensma
- Roche’s small molecule splicing modifier Evrysdi
Currently, only Biogen’s Spinraza is approved for all ages and all subtypes of SMA. For them, expansion means generating additional supporting data outside their previous clinical studies. For example, they are examining use in pre-symptomatic cases (identified by genetic screening) as well as focusing on the duration of benefit for older SMA patients. They are also exploring higher doses to see if the clinical benefit / risk is more favorable.
Novartis’ Zolgensma is FDA approved only for patients under age 2. Their challenge is that use in older patients requires intrathecal administration (vs their current IV formulation), and that until recently there was a clinical hold on that trial due to toxicities in animal studies. However, these issues were addressed. In August 2021, they were cleared to start trials for older patients (age 2-18) for intrathecal administration.
Finally, Roche’s Evrysdi is approved for all ages older than 2 months. They are currently exploring use in pre-symptomatic infants (under age 6 weeks), which could potentially expand their label to all ages.
Taken together, these expansions could open competition in both very young / pre-symptomatic patients (where all three therapies may have supporting data pending these trials) as well as patients older than 2 years old, where a successful Zolgensma entry could offer a one-time treatment option vs the ongoing Spinraza or Evrysdi regimens.
Sequenced / Combination Treatment Regimens:
In addition to monotherapy expansion strategies, the major SMA players are also exploring regimens. Because the underlying therapeutic mechanism is non-overlapping for Zolgensma (which replaces SMN1 gene expression) vs Spinraza / Evrysdi (which both rescue compensatory SMN2 gene expression), there is opportunity to explore both combination and sequencing strategies.
Figure: Ongoing and Planned SMA Sequenced Regimen Trials
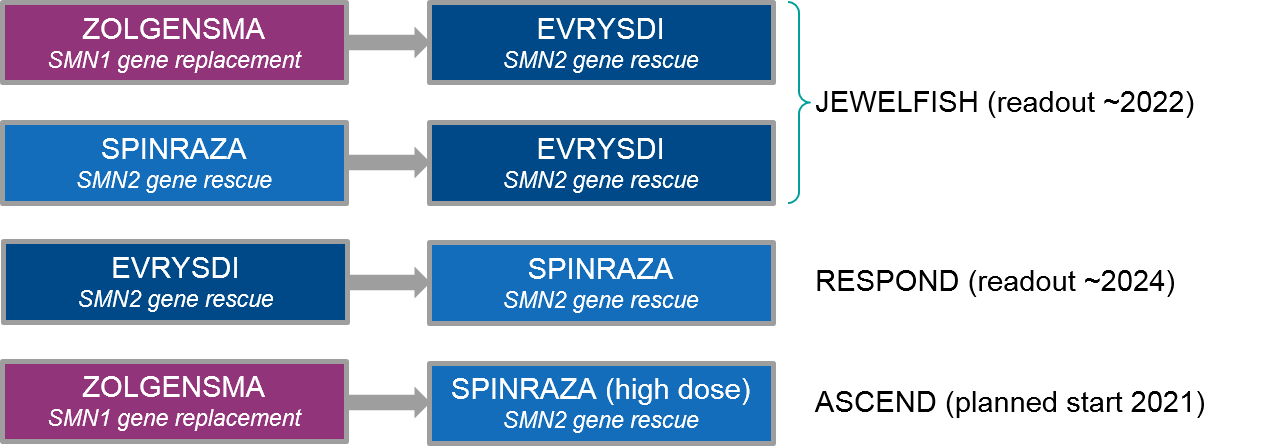
At the moment, both Spinraza and Evrysdi are in clinical trials for use after Zolgensma (and other therapies). The JEWELFISH trial is examining Evrysdi use after various other therapies. It has been enrolling since 2017 with a readout expected as early as January 2022. This trial includes prior treatment not just with the approved therapies Zolgensma and Spinraza, but also additional experimental therapies (that did not succeed), i.e., an SMN2 splicing modifier RO6885247 and a neuroprotective agent olesoxime. Preliminary data shows Evrysdi use after prior therapy increased SMN2 expression with side effects comparable to Evrysdi alone, which is encouraging for eventual incorporation into clinical practice.
The Spinraza trial (RESPOND) of patients with prior Zolgensma treatment began enrolling in January 2021 with an expected readout sometime in 2024. Because of the difference in ease of administration–Evrysdi is a daily oral medication while Spinraza is a quarterly intrathecal injection—it will be very illuminating to see how the efficacy / tolerability stacks up between these two products, and whether sequencing will improve the overall outcome. Towards this question, Biogen has recently announced a trial launch that will test whether a high dose of Spinraza will benefit patients already on Evrysdi, particularly those who have maxed out on the dose as they age.
The next question is whether it is safe and beneficial to use two products at essentially the same time. An example would be the one-time use of Zolgensma to boost SMN1 expression immediately followed by ongoing use of Spinraza or Evrysdi to target SMN2. It is possible that use of these products in combination would enable lower doses of each and therefore improve the tolerability profile. However, it is more likely that clinical practice will follow the sequential approach currently in testing, given the expense and side effect profiles of these agents. It is also quite challenging to define and demonstrate clear benefit over the already high efficacy of single agents in a combination clinical trial, which may make it difficult to successfully enroll enough participants to demonstrate additional benefit.
That said, with multiple agents in the marketplace, physicians are able to explore combination and sequencing regimens outside of clinical trials. There is at least one publication of case studies showing that introducing Evrysdi as soon as Zolgensma reaches its “plateau of therapeutic benefit” was well-tolerated and delivered “objective and subjective improvement”1.
Next-Generation / Novel Therapeutics:
The currently approved therapies are exclusively directed at boosting SMN protein production, and they do so quite effectively in most patients. That said, there are next-generation approaches for SMN boosting being developed (discussed below), as well as other targeted treatments designed to combine with SMN-directed therapies for greater efficacy.
Generally speaking, we can group novel therapies into functional classes based on how they boost SMN activity. There are:
- SMN gene-directed boosters (including the existing SMN1 replacement and SMN2 rescue therapies)
- Downstream effectors of SMN protein function that complement SMN activity.
The SMN gene-directed therapies are necessary but not always sufficient for full motor neuron protection. Future combinations involving the downstream factors may deliver even better efficacy.
Figure: Functional Classes of Existing and Emerging SMA Therapeutics

Given the underlying biology of SMN2 gene expression, namely the exon skipping and faulty protein expression, researchers have been actively exploring new approaches to rescue SMN2 gene function. The success of the existing SMN2 splicing modifier rescue therapies Spinraza and Evrysdi creates a high bar, as we see demonstrated by Novartis’ recent decision to halt their SMN2 splicing modifier (branaplam) in July 2021 due to commercial viability concerns.
Given the low likelihood of fast-follower success, it is not surprising that next generation approaches are largely focused outside the approved MOAs. For example, researchers are exploring:
- Using an antisense oligonucleotide to target a naturally-occurring long noncoding RNA (lncRNA) called SMN-AS1 that blocks SMN expression2
- Delivering a SMN2 splicing modifier (an “exon-specific U1 snRNA”) using gene therapy3
- Direct gene editing (CRISPR) of the SMN2 splice site to recover normal protein expression4
Any of these could complement Zolgensma’s SMN1 gene replacement and could possibly be used in combination with the SMN2-targeted Spinraza and Evrysdi given different MOAs (possibly allowing lower doses of each with a better side effect profile).
The approaches described above can be considered as potential replacements for existing therapies, but there is also active research and development for downstream effectors of SMN protein intended to be used in combination with existing SMN-directed therapies, typically aimed at improving muscle function. For example, Scholar Rock’s myostatin-inhibitor apitegromab has recently announced positive topline results for their Phase 2 trial indicating that addition of apitegromab to ongoing Spinraza therapy improves functional motor scores.
Another muscle-targeted therapeutic, Cytokinetics’ reldesemtiv, has been in clinical development for SMA for several years now, with promising initial results reported in 2018. However, they are currently focusing on the drug for ALS, with no trials ongoing in SMA. It is also possible that these new classes of therapy will show promise in non-SMN-driven SMA, although this population is still under-researched.
Implications for Biopharma: Competition will increase, and new product entrants will have to demonstrate clear improvement to the evolving standard of care. Novartis’ recent withdrawal of its SMN2-targeted small molecule inhibitor branaplam (due to lack of commercial viability) is an early example of the risks of fast following. For products designed to combine with (rather than replace) existing therapeutics, companies must also consider clinical trial design that enables sufficient statistical power (i.e, number of patients enrolled) to detect a potentially modest improvement over the already high standard of care. Biopharma should also consider pricing models and patient support programs for combination therapies, particularly when the combination partners are not within the same company’s portfolio and individually high-priced.
Trend 4) Increased Focus on Functional Outcomes
Until the past few years, for many patients SMA was a progressive and ultimately fatal disease with no effective interventions. Not surprisingly, the primary endpoints for the initial label-enabling trials for the now-approved therapies were focused on snapshots of motor function (using a specific assessment like independent sitting) and time to death or permanent ventilation. However even at that time, we do see interest in additional breadth of secondary endpoints that can more fully characterize the outcomes. Examples include endpoints associated with upper limb strength, feeding, and ability to walk unassisted.
More recently, we see a much broader focus on functional outcomes that include additional perspectives and measurements. For example, Spinraza’s ongoing clinical trials include parent / caregiver assessments, clinician assessment of overall change, and a quality of life assessment. Zolgensma’s ongoing trial also includes broader measures like a standard pediatric development scale and a disability inventory. Clearly, there is increasing interest in more fully characterizing the patient and parent / caregiver experience on therapy, which will in turn help regulators and treatment decision-makers generate more informed choices.
To bolster these potential subjective observations, developers are also working on more objective measures of outcomes. For example, researchers are testing the use of sensors to measure upper arm mobility5 and a wearable insole6 that will provide data on real-world gait and walking behavior for SMA patients. If validated, this type of technology could provide valuable data for future clinical development programs.
Because SMA can usually be confidently diagnosed using genetic testing and functional measures, there is typically less use of direct measures of nerve and muscle function, e.g., electromyography, biopsies. However, recent studies are looking into the use of MRI and other imaging procedures to measure disease progression and response to therapy7. Together with the traditional functional tests, imaging technologies will contribute to a more comprehensive picture of disease burden and treatment impact.
Another active area of investigation is the search for biomarkers that are predictive of outcomes on therapy. Given the high price of therapy (as well as the side effects), it is important to avoid treatment when it will not be effective. Recent studies have examined CSF biomarkers before and during Spinraza treatment, identifying several candidate biomarkers associated with disease burden and better or worse motor function outcomes to therapy8, 9.
Implications for Biopharma: At the clinical trial design stage, developers should consider a spectrum of endpoints that can be incorporated into a compelling value proposition not just for regulators but for patients and healthcare providers. That said, the overall burden on clinical trial participants as well as real-world users seeking to track their outcomes should be minimized, with a preference for non-invasive and quick or automated assessment scales. In addition, data should be gathered outside traditional clinical trials using real-world evidence, registries, and long-term follow-up of existing cohorts. Biopharma players should also consider which outcome measures will best support competitive differentiation, including safety / tolerability measures.
Learnings for Future Success
The next several years will no doubt be eventful from a research and clinical perspective, with cascading effects into the commercial arena. A major question is how current players and potential entrants can position themselves for success, given increasingly competitive pressure as well as potential pricing pressure (due to the high costs of therapy).
Another question is to what extent learnings from SMA are applicable to other disease areas. In some ways, SMA is uniquely amenable to therapeutic intervention, given the underlying biology of disease with the existing compensatory gene SMN2 available for modulation. However, the modalities used to treat SMA are certainly applicable to other disease areas that involve either loss of function gene mutations and / or gene splicing defects.
Taken together, players in the SMA space or in any similar space can consider two major categories of key success factors across the product lifecycle from early development to commercialization.
Figure: Key Success Factors

Importantly, none of these are unique to SMA. Plus, each can be applied (to varying degrees) to other rare diseases, as well as to more common ones that share similar underlying biology or product characteristics. As such, we expect more products to follow in the footsteps of SMA treatments leading to impactful therapies that can help save lives.
References:
- Oechsel KF and Cartwright MS. Combination therapy with onasemnogene and risdiplam in spinal muscular atrophy type 1. Muscle Nerve 2021 Jul 20. https://doi.org/10.1002/mus.27375
- d’Ydewalle C et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron. 2017 Jan 4; 93(1): 66–79. https://dx.doi.org/10.1016%2Fj.neuron.2016.11.033
- Donadon I et al. Rescue of spinal muscular atrophy mouse models with AAV9-Exon-specific U1 snRNA. Nucleic Acids Res. 2019 Aug 22; 47(14): 7618–7632. https://dx.doi.org/10.1093%2Fnar%2Fgkz469
- Li J et al. Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. National Science Review, Volume 7, Issue 1, January 2020, Pages 92–101, https://doi.org/10.1093/nsr/nwz131
- Annoussamy et al. Natural history of Type 2 and 3 spinal muscular atrophy: 2‐year NatHis‐SMA study. Ann Clin Transl Neurol. 2021 Feb; 8(2): 359–373. https://dx.doi.org/10.1002%2Facn3.51281
- Wearable Gait Technology Shows Promise in SMA and DMD (neurologylive.com)
- Savini et al. Pilot Study on Quantitative Cervical Cord and Muscular MRI in Spinal Muscular Atrophy: Promising Biomarkers of Disease Evolution and Treatment? Front Neurol. 2021 Mar 29;12:613834 https://doi.org/10.3389/fneur.2021.613834
- Johannsen J et al. Evaluation of putative CSF biomarkers in paediatric spinal muscular atrophy (SMA) patients before and during treatment with nusinersen. Journal of Cellular and Molecular Medicine, Open Access, 27 July 21. https://doi.org/10.1111/jcmm.16802
- Freigang et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Ann Clin Transl Neurol. 2021 May;8(5):1049-1063 https://doi.org/10.1002/acn3.51340





