
The Spinal Muscular Atrophy (SMA) treatment landscape offers a compelling case study of how very different therapeutic approaches can be successful in alleviating a devastating rare disease, leading to a complex treatment landscape with continued opportunity for both improved patient outcomes and biopharma market success. Here in Part 1, we introduce the disease and its underlying genetic cause, then discuss how the three marketed therapeutics work in unique ways to address it. Finally, we review some remaining unmet needs. In Part 2 of this series, we will describe the market trends that are moving towards addressing those unmet needs and the outlook for various players in this space.
Disease Overview
SMA refers to a collection of diseases with a common presentation, namely the progressive deterioration of motor neurons. Motor neurons are the nerves in the brain that control essential skeletal muscle movements including walking and breathing. As the nerves die, the associated muscles weaken and atrophy as well, hence the name. In its most severe form, SMA is apparent at or soon after birth and is quickly progressive. In those cases, death from respiratory failure usually occurs before age 2. However, there are multiple subtypes of the disease with different ages of presentation and typical outcomes.
Table: SMA is categorized into multiple types, based on the age of presentation and severity of symptoms
| Subtype | Age at Onset | Symptoms | Outcome & Impact |
|---|---|---|---|
| Type 0 (prenatal) | Prenatal / fetal | Severely reduced or no motor function (requirement for ventilation) | Death in utero or before 6 months |
| Type 1 (I; infantile-onset) | <6 months | Hypotonia, diminished movements / reflexes, feeding and breathing difficulties | Without treatment, death before age 2 due to respiratory failure |
| Type 2 (II, intermediate) | 6-18 months | Can sit but not walk, may have respiratory difficulties | Variable progression with reduced life expectancy (e.g., early adulthood) |
| Type 3 (III) | After 18 months | Can walk with difficulty, scoliosis and joint contractures | Increased risk of respiratory illnesses but normal lifespan |
| Type 4 (IV) | After 21 years | Mild to moderate proximal muscle weakness | Normal lifespan |
Across the most common types (Types 1-3), the total incidence is estimated at 8-10 / 100,000 births.1 Type 1 is the most common, making up about 60% of cases, with Type 2 and Type 3 making up about 20% each. Type 0 is difficult to quantify as many affected individuals die in utero or at birth, and Type 4 can have mild symptoms and therefore be under-diagnosed.
SMA is a genetic-driven disease, with the majority of cases (~96%)2 due to mutations in the gene SMN1, which encodes a protein called SMN (“Survival Motor Neuron”) that maintains motor neuron health and survival. If either of the two copies of this gene in the genome is functional, it is sufficient for SMN protein production and there is no disease. However, if both gene copies are defective, then there is insufficient production of SMN and motor neurons will die off, leading to SMA.
If SMA is driven by a single gene, then why are there different subtypes with variable ages of onset, symptoms, and courses of the disease? This is because our genomes also carry a “back-up” gene called SMN2, which can partially compensate for mutated SMN1. The SMN2 gene sequence is extremely similar to SMN1, and therefore also produces the SMN protein. However, it does so at much lower levels due to a genetic variation called “exon skipping” that results in frequent errors of protein production.
Normally, when a gene expresses a protein, it makes an RNA copy that then undergoes an intermediate step called “splicing” where segments of the gene copy are removed and the remaining pieces (the exons) are stitched back together before being translated into a protein. The relevant splicing error in SMN2, “exon skipping”, occurs when a DNA signature within the gene triggers this machinery to discard that exon rather than including it in the final version for protein production, which therefore ruins the final protein. Exon skipping does not happen 100% of the time however, so some small percentage of SMN2 gene copies are stitched together correctly and result in accurate SMN protein production.
Figure: SMA is caused by inadequate SMN protein production, with SMN2 gene expression being insufficient to compensate for SMN1 gene dysfunction

Another key source of variation between patients is that for some individuals, the genome has more than 2 copies of SMN2, enabling higher overall production of SMN protein. The number of SMN2 copies is therefore inversely proportional to the disease severity. Individuals with Type 1 SMA typically only have 2 SMN2 copies whereas those with Type 2-4 SMA are likely to have more, thus allowing more compensatory expression of SMN protein (and less severe disease).
While the above explanation may seem quite “in the weeds”, it’s necessary to understand the two major mechanisms of action (MOAs) of the approved SMA therapeutics, namely SMN1 gene replacement and SMN2 gene rescue, and how they may complement each other to boost SMN protein production.
Figure: SMA therapeutic approaches address independent pathways, suggesting opportunity to combine or sequence
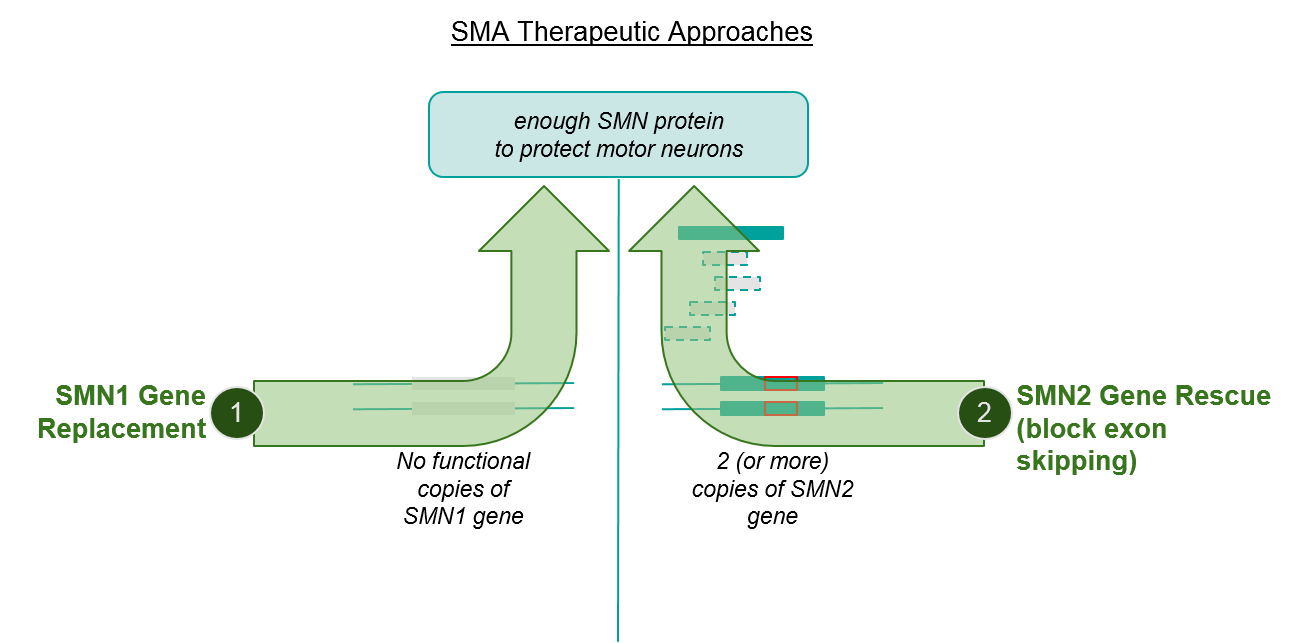
Importantly as we consider therapeutic approaches, not all SMA cases are driven by an SMN1 mutation. Other driver genes have been identified including VAPB, DYNC1H1, BICD2, and UBA1. We will discuss further when we describe remaining unmet needs in this disease space.
Therapeutic Landscape
Given that the vast majority of SMA cases are caused by insufficient production of the SMN protein, this has been the focus of therapeutic development. SMA now has three clinically effective and approved therapeutics that each use different methods to recover SMN protein production.
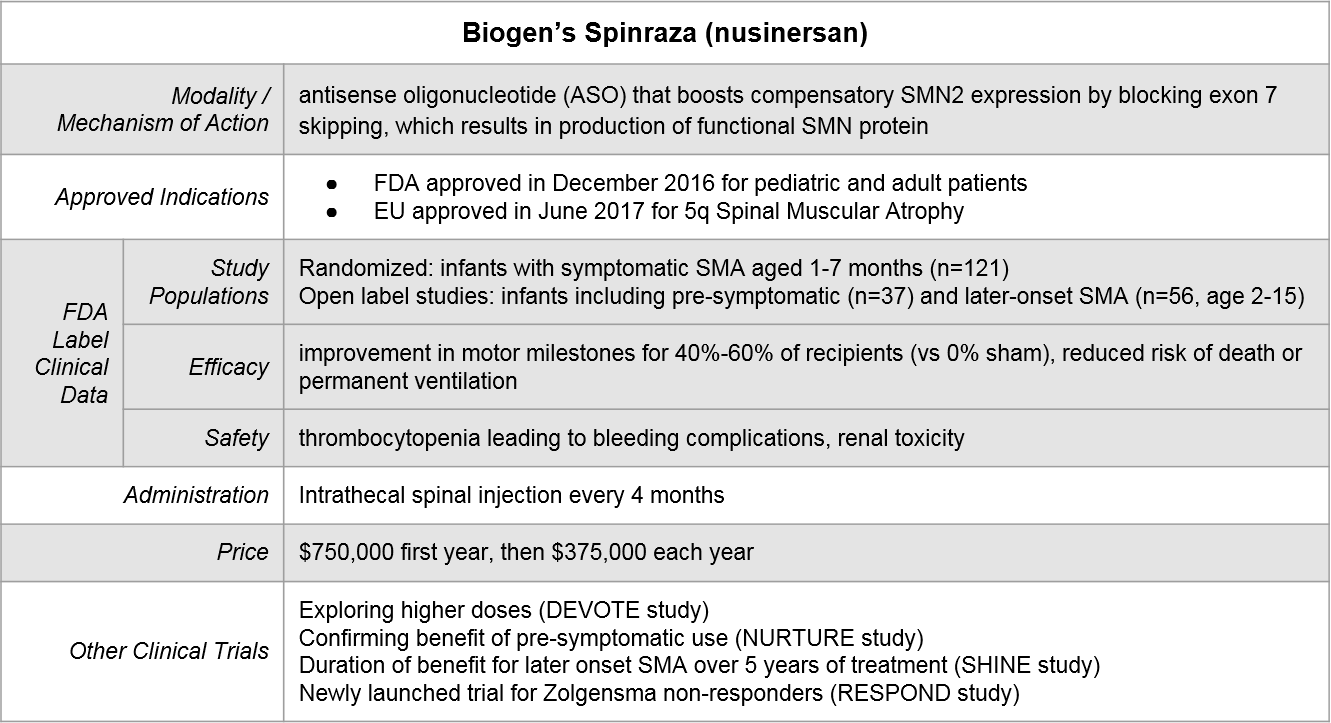
Biogen’s Spinraza® was the first SMA treatment to be FDA approved, in December of 2016. It takes an SMN2 gene rescue approach by blocking SMN2 exon skipping (which results in recovery of functional SMN protein expression). After an initial dose-dense regimen, Spinraza is delivered every 4 months via intrathecal spinal injection.
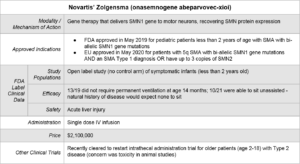
Novartis’ Zolgensma® was FDA approved May of 2019. Unlike Spinraza which uses a novel modality (antisense oligonucleotide), it takes a more established rare disease approach of gene therapy: introducing a functional SMN1 gene into the motor neurons using a adenoviral vector.
Roche’s Evrysdi® is the most recently approved by the FDA (August, 2020). It works similarly to Spinraza but uses a different modality. It is a small molecule inhibitor and is a daily oral medication.

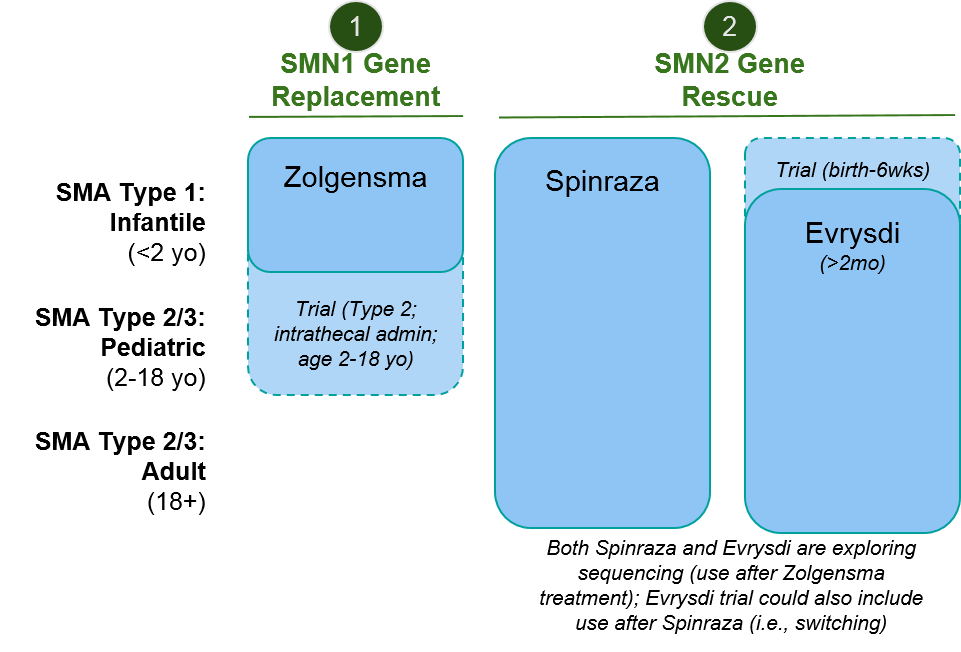
Because of variations in both label language as well as supporting clinical data, there is partial but not complete overlap in where each of these therapeutics can play. Importantly, both Spinraza and Evrysdi have initiated clinical trials to explore sequencing of their therapeutics after Zolgensma. The eligibility language for the Evrysdi trial also encompasses prior use of Spinraza, allowing for the possible exploration of switching.
Figure: Approved Indications & Ongoing Exploratory Trials
Unmet Needs
Despite the incredible achievements of successfully developing and launching these three therapeutics, there is still significant unmet need for SMA patients:
- Earlier detection and intervention: SMA is recommended as part of newborn screening panels, but not all newborns receive screening. There are also age limitations on approved therapeutics which limit the window during which therapy can be used.
- Non-SMN driven disease: The available therapeutics would not be expected for work for those ~6% of patients whose disease is driven by a non-SMN mutation.
- Better efficacy: While all three therapeutics offer impressive reductions in death and improvements in motor function, not all patients benefit equally. There is still work to be done in identifying biomarkers and other predictors of response, and in developing new regimens (combinatorial or sequenced) and / or new therapeutics to improve outcomes for those for whom monotherapy is less successful.
- Better access: These therapeutics are not yet approved worldwide. Where they are approved, cost is a concern as well. Zolgensma has the dubious distinction of being the most expensive medication in the US, and the other two medications are also costly.
Conclusion
Over the past 5 years, SMA went from untreatable (and fatal for Type 1) to a more chronic condition that can be managed with a choice of one of three effective therapeutics. Each of these three therapeutics successfully leveraged a unique therapeutic approach, leaving room for beneficial combinations as well as providing a template for other genetic diseases with opportunity for exon skipping rescue (e.g., in DMD to rescue dystrophin, FTDP-17 to rescue MAPT, in familial dysautonomia to rescue IKBKAP).3 The progress to date for SMA has been rapid and significant, leaving us hopeful for a future where the remaining unmet needs are addressed as well.
In Part 2, we will dig deeper into how key players in this market are meeting the challenges described above, and explore the near-term outlook for the SMA landscape overall.
Notes:
[1] Verhaart, I.E.C., Robertson, A., Wilson, I.J. et al. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 12, 124 (2017). https://doi.org/10.1186/s13023-017-0671-8
[2] Ibid.
[3] Tang, Zhichao, Junxing Zhao, Zach J. Pearson, Zarko V. Boskovic, and Jingxin Wang. 2021. “RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays” Molecules 26, no. 8: 2263. https://doi.org/10.3390/molecules26082263





