
In Part 1 of this series, we reviewed recent developments and upcoming milestones that are shaping the cell-based therapy landscape. Here in Part 2, we explore the various factors and challenges that will influence how each cell-based therapy—and the class as whole—will perform in the marketplace over the next few years. Some of these factors are unique to each specific therapy (e.g., design decisions that influence market success), some are intrinsic to the class (e.g., manufacturing and supply chain complexities, commercialization), and some are extrinsic (.e.g., out of class disrupters, coverage and reimbursement landscape).
For purposes of this paper, we define cell-based therapy classes as follows:
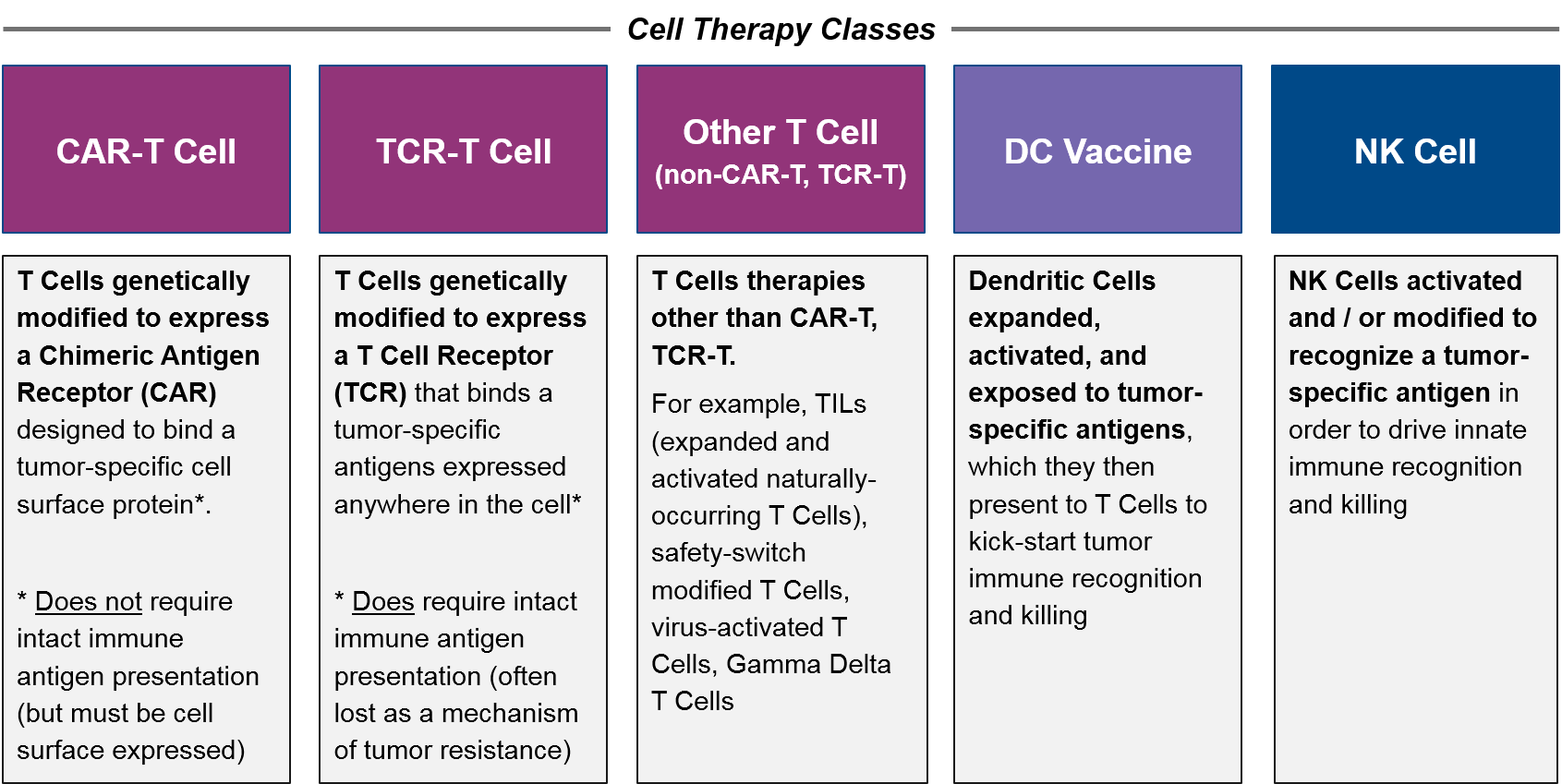
Comparison between allogeneic CAR-T Cells and autologous CAR-T Cells is therefore “intraclass”, whereas comparisons between CAR-T Cells, TCR-T Cells, and other T Cell-Based Therapies are “interclass”. Out-of-class comparators are any other cancer therapy, but the most relevant competition are other immunotherapies such as checkpoint inhibitors, T Cell / NK Cell Engagers (bispecific antibodies), and cancer vaccine approaches.
Differentiation Opportunities
To differentiate and succeed from a product attribute perspective, each new cell-based therapy must achieve one or more of the following efficacy and / or safety objectives:

Strategies for breakthroughs or expanding indications in both liquid and solid tumors will include efficacy and safety modifications of existing therapies plus different positioning (patient selection, other lines of therapy, combination regimens). In addition, new targets and design enhancements will enable breakthroughs in liquid tumor types beyond the currently established Multiple Myeloma, NHL, and B-ALL.
For solid tumors, the near-term opportunity will be in unlocking the efficacy of known targets in any solid tumor (as none have shown clear sensitivity to cell-based therapies). This will no doubt require efficacy modifications and / or combination strategies that address solid tumor barriers like physical access to the tumor as well as tumor-mediated immune suppression.
Once a cell-based therapy shows efficacy in an indication, the question becomes how to address whatever percentage of recipients do not respond. In practice, this means we must address two distinct forms of resistance to maximize clinical benefit – primary and secondary resistance. For B-ALL and DLBCL, roughly 20% and 50% of recipients (respectively) do not achieve remission with CD19 CAR-T (i.e., the cancer is resistant from the start). This “primary resistance” is associated with depletion of infused T Cells and is assumed to be due to inherent defects in the recipient’s modified T Cells.
We therefore expect the use of more standardized allogeneic (donor-derived) cell products to address at least some of this primary resistance. Recent research has also indicated that primary resistance may come from the tumor as well, via suppression of the molecular pathways that drive immune-mediated tumor cell death (apoptosis). Exposure to these death-resistant tumor cells may be “exhausting” the T Cells, leading to the depletion noted above. It may be that these resistance mechanisms will need to be targeted to fully address primary resistance
For those recipients who do respond well to cell-based therapy but whose cancers ultimately progress, the mechanism of this “secondary resistance” is typically antigen loss. This means that the tumor cells stop expressing or presenting the targeted antigen and therefore become undetectable to the T Cells.
Tumors have such variable underlying mutations that it is challenging to envision a targeted cell-based therapy strategy to address secondary resistance. Therefore, the focus has been on stopping it before it starts by more efficiently killing the original tumor cell population. For example, some drug developers are already exploring dual targeting therapies which could help address this resistance before it begins. Additionally, combination strategies of cell-based therapies plus targeted drugs or systemic therapies could be helpful to stop this form of resistance before it takes hold.
Challenges Overview
Besides achieving regulatory approval based on differentiation from standard of care on the basis of efficacy or safety, cell-based therapies must also succeed in a complex external environment that includes manufacturing challenges, regulatory and pricing pressure, and intensifying competitive pressure.
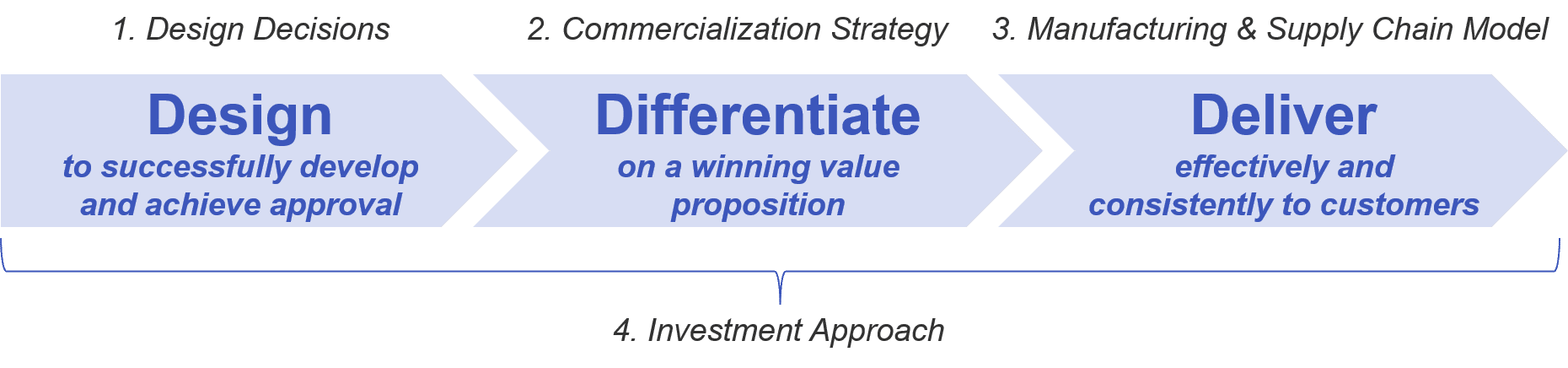
The successful cell-based therapy must therefore navigate the following challenges:
- Design Decisions:
- Cell type and source
- Target (if any)
- Genetic modifications (e.g., targeting, safety edits)
- Genetic modification method
- Diagnostics (as applicable)
- Commercialization Strategy:
- Optimized commercial model
- Value proposition to address anticipated coverage and reimbursement challenges
- Manufacturing and Supply Chain:
- Optimal balance of standardization and efficiency
- Investment Approach:
- Products and / or capabilities required
- In-house vs partnering, acquisition, etc
The ultimate goals of approval and market success will depend on the factors above, but also to what extent they position the therapy within the competitive landscape:
- Differentiation from intra/interclass cell therapy competitors:
- Allogeneic vs autologous
- CAR-T vs TCR
- NK vs T cell-based
- Differentiation from out-of-class potential disrupters:
- T Cell / NK Cell Engagers
- Cancer vaccines
- Other immunotherapy combos
Design Decisions
Cell-based therapy design is a critical and foundational piece of the puzzle, guiding the differentiation strategy by setting the bounds for efficacy, safety, and manufacturing feasibility for each product. At the beginning of each cell therapy program, developers must make numerous design decisions that should solve for one or more of the following product attribute differentiation strategies, addressing known or suspected efficacy, safety and / or manufacturing challenges:

From an efficacy perspective, the key decisions are cell type, target selection, and modification design including both targeting modifications (like CAR or TCR) as well as secondary activating modifications to boost function. For most targets, a companion diagnostic will be required to identify the most likely responders (notable exceptions are CD19 and BCMA in NHL and MM, respectively, which are highly expressed across all cancer cells).
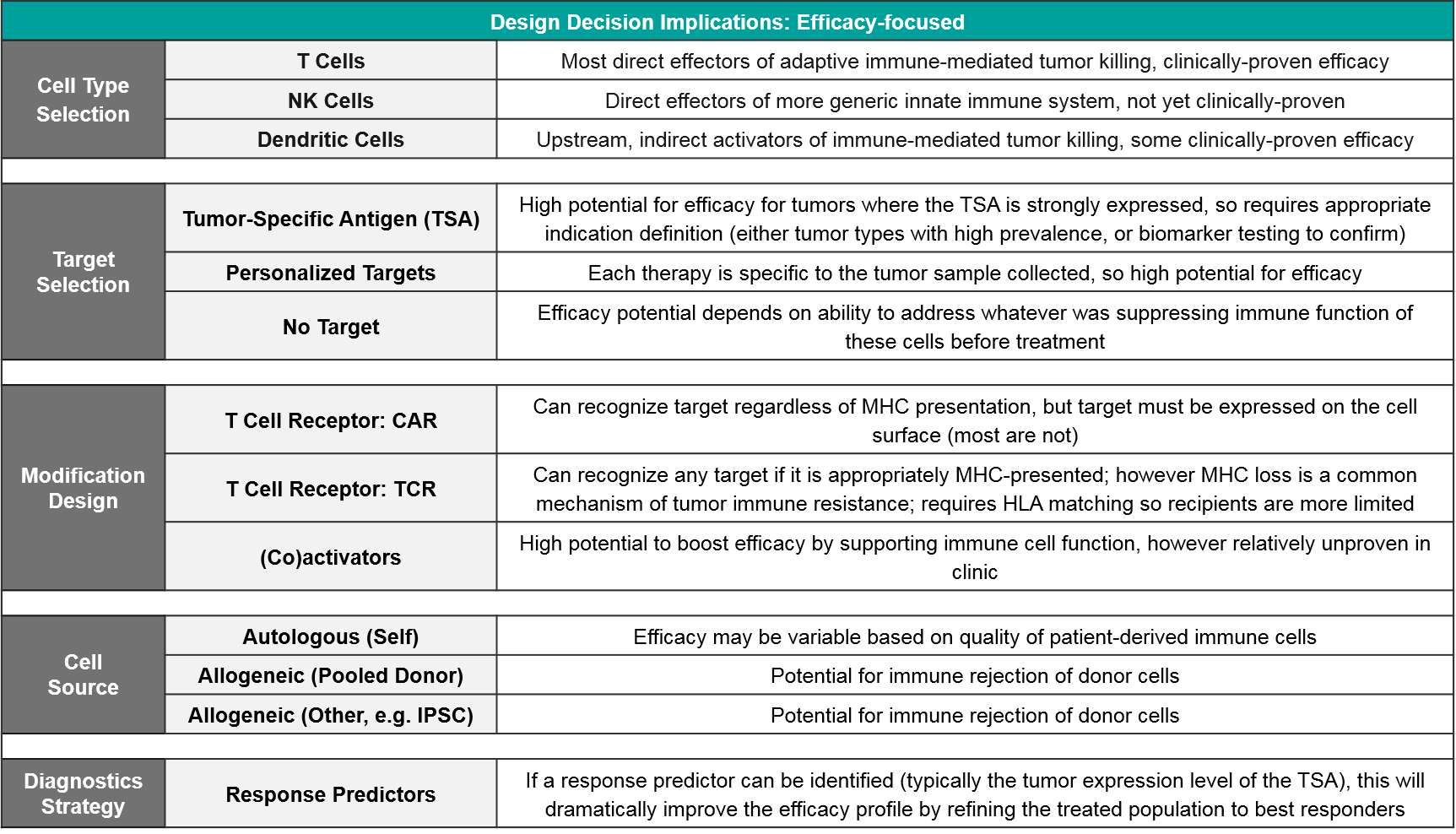
Of the three cell types, T Cells have both a solid scientific rationale (they are the direct effectors of immune-mediating killing) as well clinically proven efficacy. NK Cells also have a solid rationale (they are direct effectors of another type of immune-mediated killing) but they are not yet clinically proven. Dendritic Cells act in a different, indirect way, by priming the existing T Cells to react to tumor antigens. Their clinical efficacy is fairly well studied and is more mixed than that of T Cell-based therapies.
From a target perspective, while target selection is the best way to direct immune-mediated killing, it has two important considerations:
- Given the heterogeneous nature of tumors, the selected target will typically not be expressed by all tumor cells. This results in both primary and secondary resistance as the non-expressers evade the therapy and then spread throughout the body. Depending on the prevalence of target expression within an indication, a companion diagnostic strategy should be considered.
- If the selected target is expressed on healthy cells, or if healthy cells express a target similar enough to trigger recognition, there is also a risk of off-target activation which can result in serious Adverse Events including death.
However, if no target is selected (e.g., for something like TILs where the existing immune cells are simply expanded ex vivo), the efficacy upside may be lower as these cells may not recognize the tumor cells, or they may be suppressed by the tumor microenvironment.
Modification choice between CAR and TCR is a critical component of efficacy and it is intertwined with target selection. This is because CAR and TCR each have specific abilities and limitations for immune recognition. TCR is a slightly modified version of the endogenous T Cell Receptor. It works via the same mechanism of recognizing antigens that are gathered from anywhere in the cell and then presented at the cell surface by MHC molecules. TCR efficacy can be therefore limited by MHC loss which is a common mechanism of tumor immune resistance. CAR do not require MHC presentation and instead directly bind any cell surface-expressed molecule. If the desired target is not expressed on the tumor cell surface, then CAR is not an option. In addition to CAR and TCR, there are modifications intended to boost immune activity, like co-expressed stimulatory molecules.
While certain cell types, targets, and modifications are known to have higher potential efficacy, the inherent trade-off is that the more effective a cell therapy product is, the higher the risk of either off-target immune activation or immune overreaction (Cytokine Release Syndrome).
Therefore, safety implications are critically-important factors for design decisions. For both cell type selection and target selection, the safety risks are typically proportional to the anticipated efficacy. Many pharma and biotech companies are including safety-focused modification designs to their newer products to address these risks. Examples include safety edits that remove extraneous TCRs which could lead to Graft vs Host Disease, and safety switches that enable physicians to suppress or eliminate cell-based therapies that are triggering a severe Adverse Event. A related consideration is histocompatibility testing – for non-patient-derived (i.e., allogeneic) cell therapies that are not safety-edited, HLA matching must be done to minimize the risk of immune reaction between the donor and host cells.
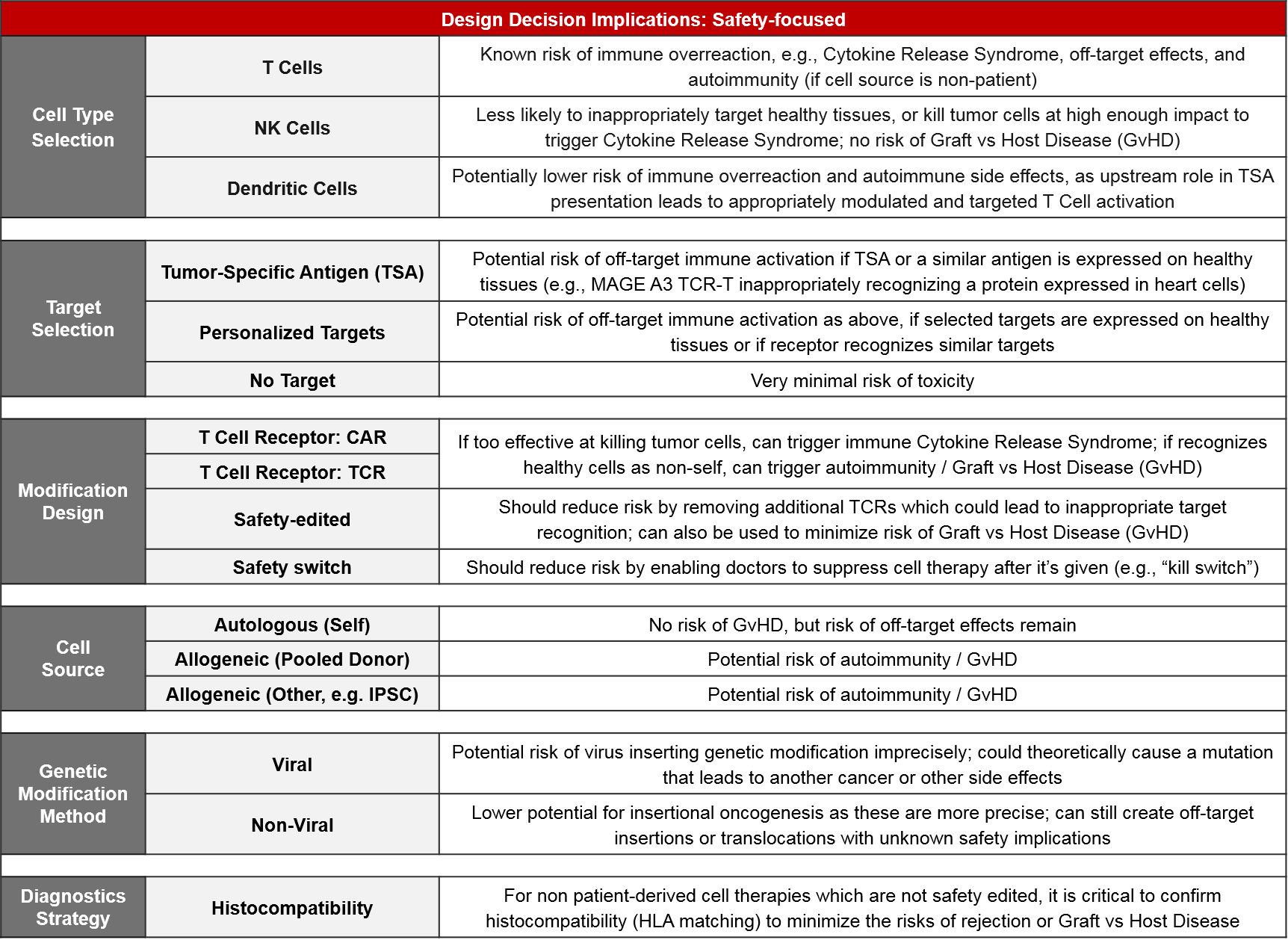
Finally, in addition to defining a cell therapy design that appropriately balances efficacy and safety, the pharma or biotech company must also account for feasibility impacts on manufacturing and cost / price.

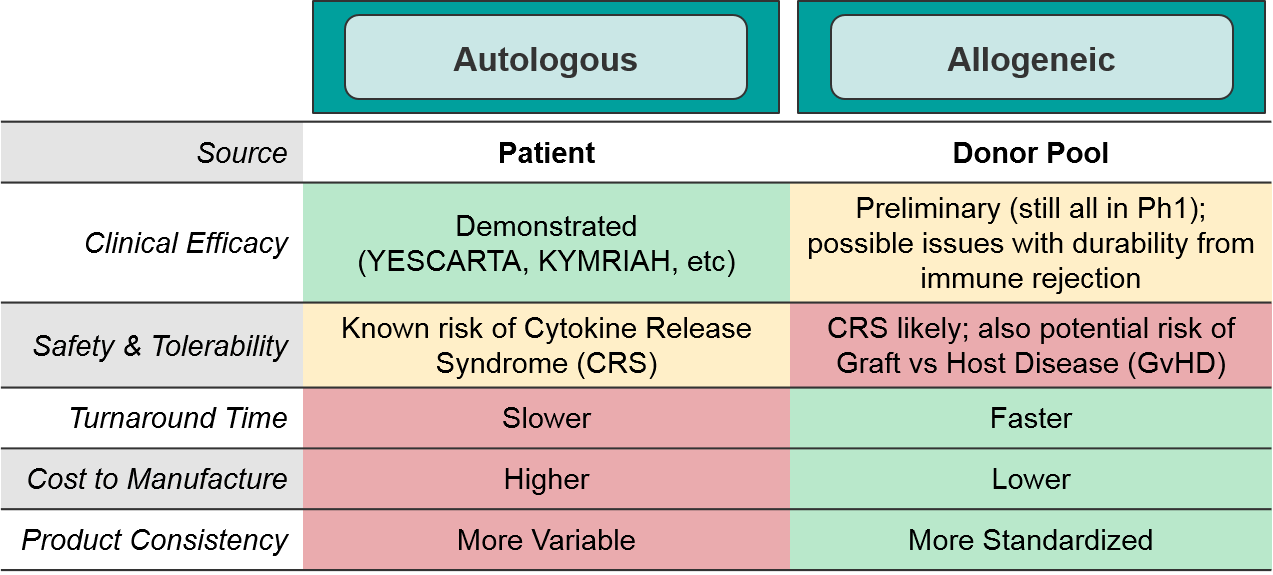
Here too, there are trade-offs between ease of manufacturing and safety risks. Namely, the newer cell therapies in development are primarily allogeneic because it is much cheaper and standardized to use a more consistent source, but there are potential safety risks from donor/recipient immune reactions. To date, the anticipated safety issues with allogeneic-sourced therapies (namely Graft vs Host Disease) have not occurred. Similarly, the potential risk of insertional oncogenesis from imprecise viral modification methods has not been observed in the clinic.
Commercialization Strategy
As a cell therapy is designed and moves through the clinic towards approval, the company must develop and refine a value proposition to build its differentiation strategy from. Because cell therapies are high-priced, and often have serious potential side effects, clarifying the value proposition is crucial. Defining the appropriate patient population (i.e., those most likely to have a beneficial response) sets the stage for the value proposition which in turn underpins regulatory approval and the eventual pricing model. Given the relatively small size of early stage trials which will inform registrational trials (which also may have relatively low numbers), it is essential to set up trials for success from the beginning with likely responders.
As with other immunotherapies, cell-based therapies typically have a subset of deep responders. Also similar to other immunotherapies, we have not yet fully defined predictive biomarkers for many cell-based therapies. For those that are Tumor Specific Antigen directed, like the CD19 CAR-T or BCMA CAR-T, the known or inferred expression of that marker on the tumor cell acts as a mostly predictive biomarker. But still, a subset of those tumors do not respond to the cell therapy, and biomarker-based identification of those non-responders would save those patients from the risks of cell therapy treatment, and potentially open new avenues of investigation for future therapies. For those cell-based therapies that target TSAs expressed across multiple tumor types and demonstrate the associated efficacy across tumor types, a tumor-type agnostic strategy may be considered to maximize both regulatory approval and reimbursement. In any setting where a TSA is less than ubiquitously expressed, a companion diagnostic strategy will be required.
Once the patient group or indication and associated therapeutic benefit is well-defined, the pricing model can be generated. Because the therapeutic benefit of cell-based therapies can be somewhat “all or none”, some manufacturers are exploring a “pay for performance” model where customers only pay the high price if the therapy works. For example, Novartis launched KYMRIAH with an outcomes-based model that required the product to meet specific targets to be eligible for reimbursement. Another possible model is annuity-based, where a payer reimburses over time based on continued good outcomes.
In addition, companies are also exploring patient access and financial assistance programs. For example, Novartis just recently unveiled a Travel Assistance Program for KYMRIAH. This program will assist low-income recipients with travel, lodging, and other out-of-pocket expenses required to obtain treatment at one of the 100 designated centers within the US.
The relationship between value and pricing is critical because it factors heavily into coverage and guidelines inclusion decisions. For example, both Gilead’s YESCARTA and Novartis’ KYMRIAH were initially rejected by the UK’s approval body NICE, citing failure to meet cost-effectiveness thresholds. Ultimately, both therapies were approved for reimbursement by NHS after negotiating confidential price cuts.
In addition to cost-effectiveness, manufacturing consistency is a major factor. Novartis has been plagued with issues in manufacturing KYMRIAH to FDA standards, resulting in a loss of profit as they cannot collect payment for these “out of spec” products, despite emerging evidence that they can still be clinically effective.
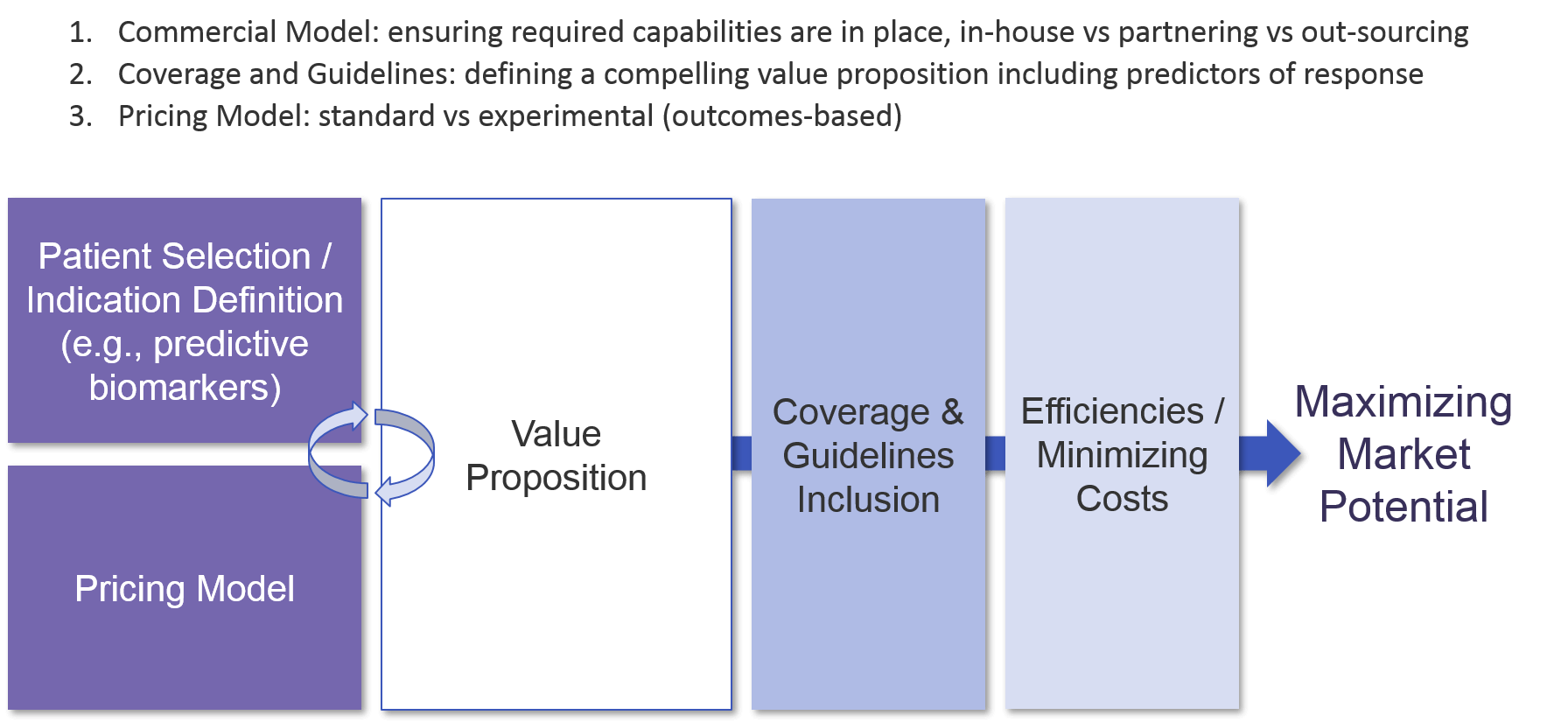
Finally, once the therapy is introduced to the market, it is critical that patients can easily access the product. One option is to focus on select Centers of Excellence (COE) before expanding towards a more decentralized model that delivers to broader point of care sites. Each company must decide to what extent they will shape the market by creating these new COEs vs leveraging existing opportunities. Providers at COEs who are already familiar with cell therapies will need some initial education and support based on the new product attributes, whereas providers who are completely new to cell therapies will require significantly more investment to ensure they can successfully deliver the therapy, confidently handle potential adverse events, etc. An upfront market landscape assessment that accounts for existing expertise (current clinical trial investigators, COEs for the competition) as well as patient geographies and supply chain considerations will enable a robust strategy on where to start and how to expand the footprint over time. An early decision on a specific COE strategy will also inform the supply chain design so that it can meet the anticipated needs of these providers. Conversely, supply chain considerations may bound the COE approach, for example if certain specific locations are difficult to reach.
Manufacturing and Supply Chain Optimization
Cell therapies are challenging to manufacture and keep consistent to regulatory standards. Unlike small molecule or antibody-based therapies, they are live cells that must be maintained over time and may come from variable sources (e.g., individual patients, donor pools). As mentioned above, Novartis’ already approved CAR-T therapy KYMRIAH has been challenged by manufacturing issues off and on since its launch in 2017, namely being unable to produce enough cells for certain patients.
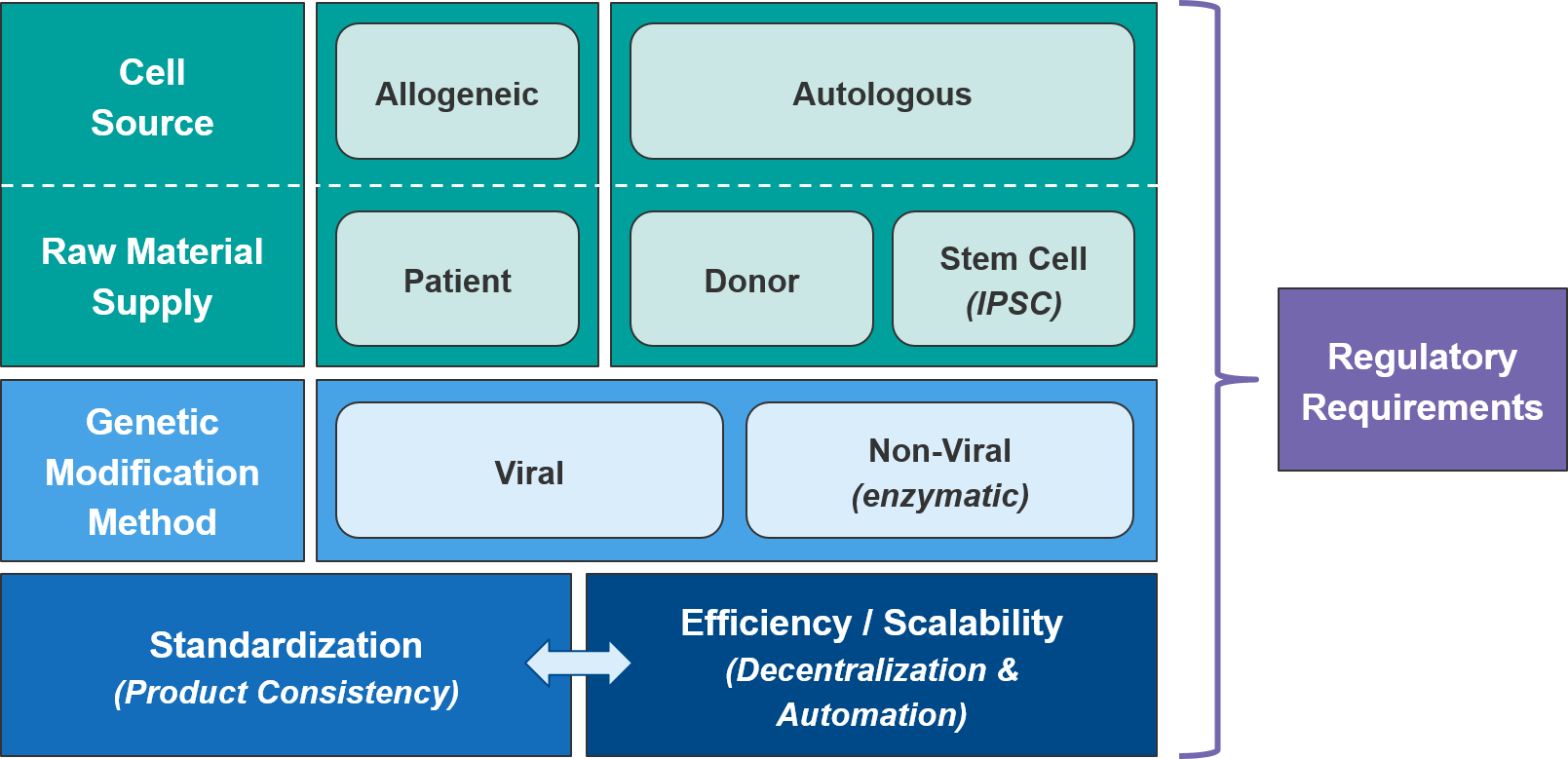
As a class, autologous (patient-derived) cell-based therapies are particularly challenging to manufacture, as there is significant variation between patients and the starting material of their harvested immune cells. As the industry moves towards allogeneic (non-patient-derived) therapies, we expect manufacturing challenges to ease somewhat as the cell sources become more standardized.
Within the allogeneic category, there are two options for cell sourcing: first, a donor-derived pool of cells and second, an artificially generated stem cell population from which immune cells can be derived as needed. The latter category, also called “IPSC” for “Induced-Pluripotent Stem Cell” would be cheaper and likely more consistent. But IPSC-based therapies are only just now reaching the clinic so we will be watching closely for proof of efficacy and safety. However, regardless of source, manufacturers will still need to manage their raw material supply, as living cells are much more difficult to maintain and transport than are small molecule or antibody-based therapies.
The type of genetic modification method also impacts manufacturing. The more established viral methods are efficient at transfection (delivering the genetic modifications into the target immune cells), but their scalability is somewhat limited and there is a perceived risk associated with their more imprecise gene insertion. Of the non-viral methods, the most clinically advanced are the DNA editing enzymes such as the transposases or CRISPR endonucleases. These enzymes target specific DNA sequences and swap in the desired genetic modification in a highly precise manner. While these enzymatic methods are more scalable, they also have a lower transfection efficiency and are not yet clinically proven.

As the cell therapy moves from pre-clinical testing into the clinic, quality control and standardization become critically important. This is particularly challenging for autologous cell therapies as each patient’s immune cells may be of differing quality. The simplest solution for standardization is to centralize manufacturing, but that comes at significant cost to efficiency and scalability. Manufacturers can improve efficiency and reduce capacity constraints by working with CMOs with cell therapy expertise. However, these savings come at some risk of quality control and lack of competitive differentiation (the CMOs can work for competitors as well, using the learnings achieved from prior work). Regardless of the specific delivery model, the standardization vs efficiency trade-off can be significantly relieved by process improvements, especially automation.
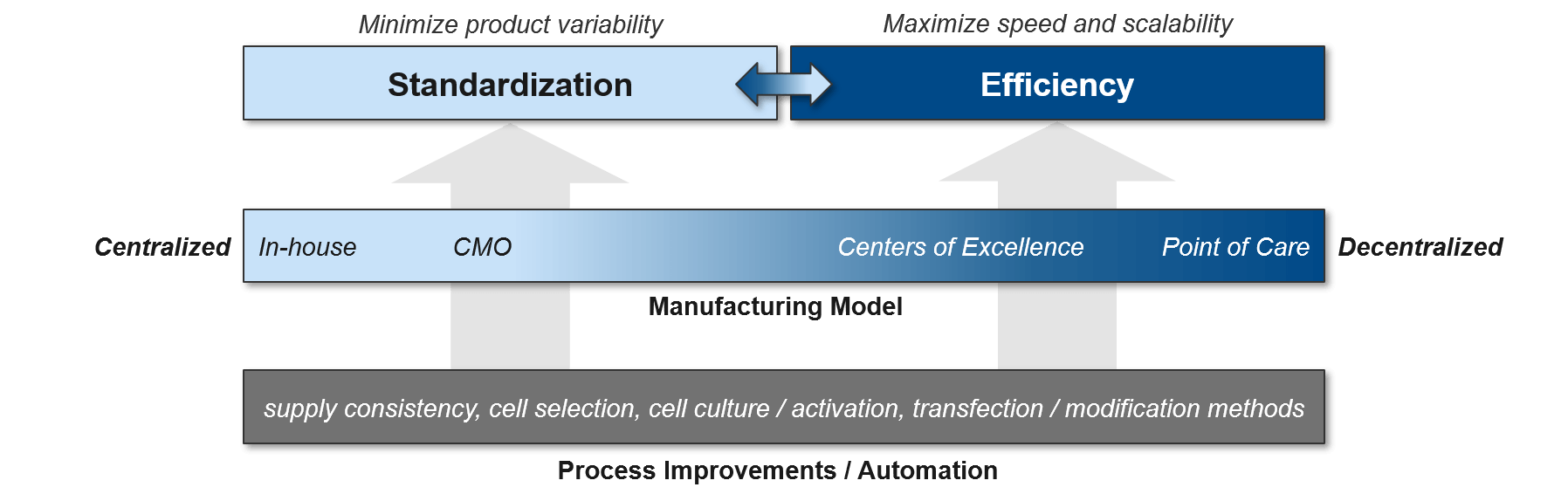
Currently, cell therapy production is quite manual and laborious, with opportunities for improvements throughout the process (beginning with the initial population of cells and extending through the product-specific modifications that will ultimately generate the final product). Shifting both the cell selection and cell modification processes towards automation will improve both standardization and efficiency.

Finally, manufacturing must meet all regulatory requirements for the FDA, EMA, or whatever regulatory body manages the geography in which they will be selling. This may include generating new supporting data. For example, Novartis recently presented data showing that “out-of-spec” KYMRIAH batches still delivered clinical benefit, setting the stage for them to negotiate relaxed FDA requirements and broader reimbursement.
Investment Requirements and Approaches
To successfully develop and sell a cell therapy, the manufacturer must have a model in place to develop and maintain delivery of each cell therapy product. Companies must be prepared for both upfront and ongoing investment to successfully design and launch the cell therapy, effectively differentiate it in an increasingly value-driven marketplace, and effectively deliver it to customers over time. These investments are driven not just by the underlying design of the therapy, but by evolving pressures from competitors and an increasingly value-driven reimbursement landscape.
As a company prepares to bring a new cell therapy to market, it must decide whether the investment will be directed in-house or outwards towards partnering or acquisition strategies. This decision applies to both specific assets as well as general capabilities. For companies who are expanding their portfolios beyond the traditional small molecule or antibody-based therapies into cell-based therapies but wish to do it solely in-house, there will be significant upfront investment in getting the appropriate capabilities in place. Key examples include genetic modification methods (viral or enzyme-based) and production of live cell-based therapies, including maintenance and transportation of these living cells to the point of care.
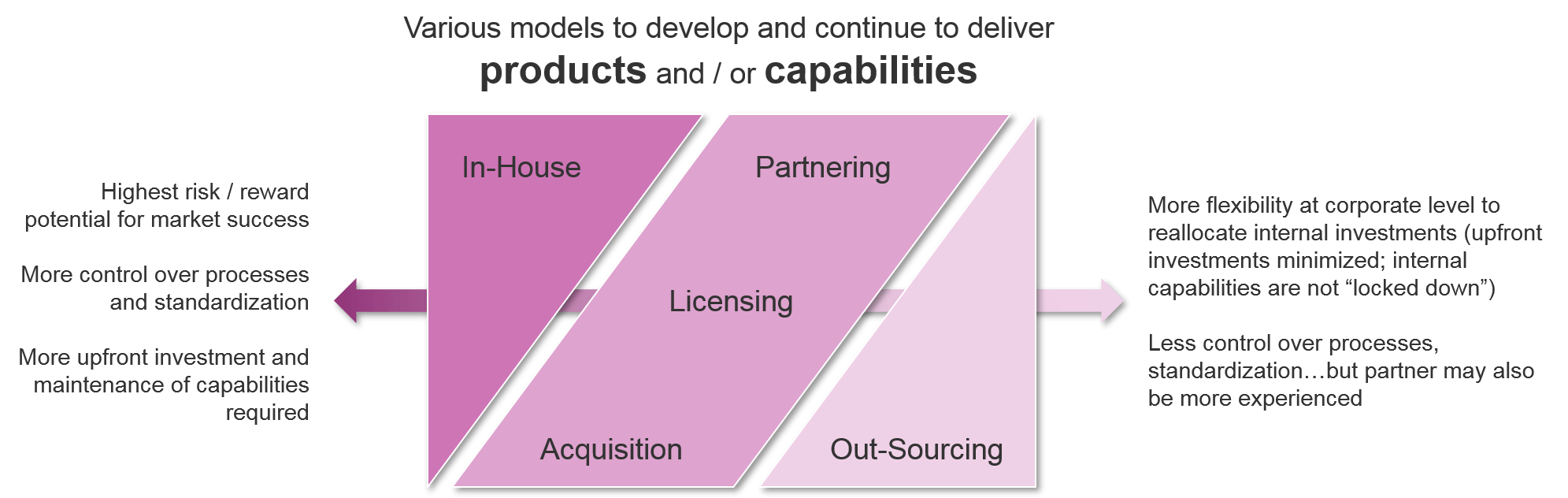
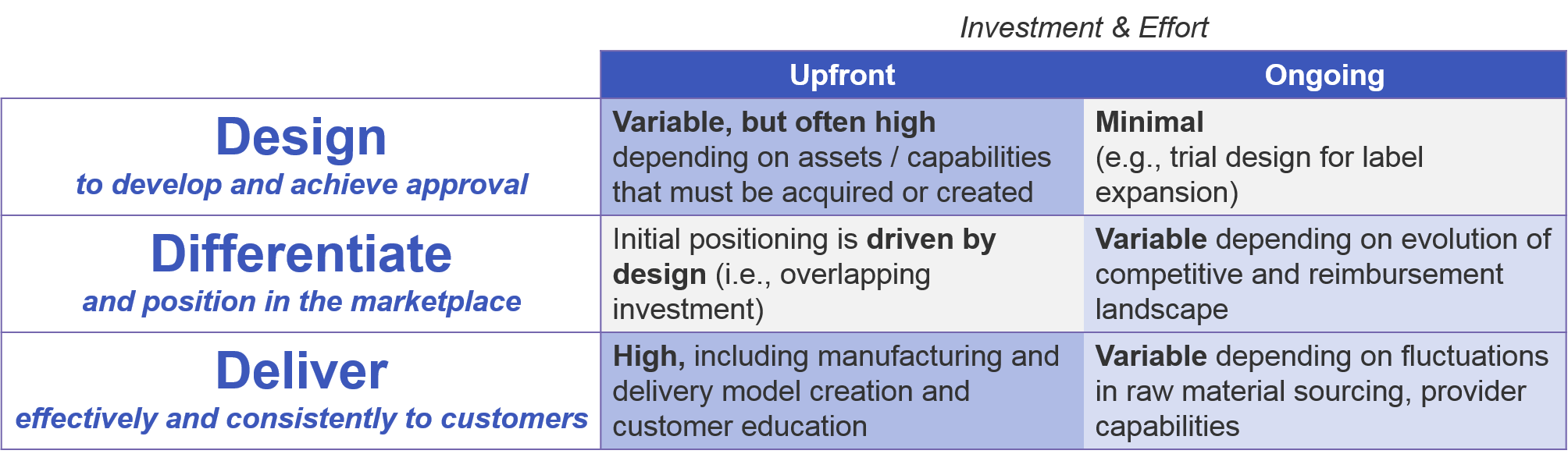
As compared to more traditional therapies, cell-based therapies require both greater upfront and ongoing investment. This is due to the complexity of design, which requires expertise in genetic engineering and live cell production, as well as the complexity of the manufacturing and delivery, which is subject to fluctuations in supply of living cells. Additionally, companies must ensure ongoing education of providers to successfully deliver a therapy with which they may have minimal experience.
Many major players in the cell therapy space are blending their investment approach, building out their portfolio with a mix of their internal pipeline as well as licensed or partnered assets, and building out their capabilities with a mix of internal investment, corporate acquisitions, and out-sourced manufacturing. For example, within the past few years various companies have bolstered their cell therapy portfolios and capabilities in the following ways:
- Gilead acquired Kite Pharmaceuticals, including their portfolio of CAR-T and TCR assets
- BMS acquired Celgene, including their portfolio of CAR-T
- Allogene obtained rights to CAR-T assets that Pfizer had licensed from Cellectis in return for a percentage stake in ownership of Allogene
- Celgene and Bluebird Bio are partnering to co-develop various cell and gene therapies
- Celyad licensed their CAR-T design to Novartis in return for upfront and milestone payments
- Sangamo and Kite/Gilead are collaborating to use Sangamo’s gene editing platform in return for upfront and milestone payments
Because the upfront investment is so high, the decision to develop in-house or acquire another company’s assets and / or capabilities becomes more appealing if the company has a long-term interest in developing additional cell therapies beyond their lead asset. If a company is primarily seeking to diversify targets or indications in a situation where a cell therapy is the best fit, then a more “hands off” approach of partnering and / or out-sourcing specific capabilities may make better sense. Companies must also carefully consider how to ramp up over time without risking a failure to deliver, while also not overinvesting upfront with associated operating losses.
Competition: Between Cell-Based Therapies
Within the cell-based therapy landscape, there are several key competitive battles shaping up. In addition to the expected in-target competition for CD19 and BCMA autologous CAR-T, there are also allogeneic competitors for both targets expected to release Ph1 data within the next year or so. These data will be critical in shaping the future path of the industry – either inevitably towards the cheaper, easier to produce “off the shelf” allogeneic therapies, or if those prove less clinically efficacious, remaining with the personalized autologous cell therapies. Finally, academic centers are exploring innovative approaches in cell therapies. Should those prove viable, academic centers would seek to partner with corporate manufacturers to commercialize, creating both opportunities and threats for existing players.

Competition: Out-of-Class Potential Disruptors
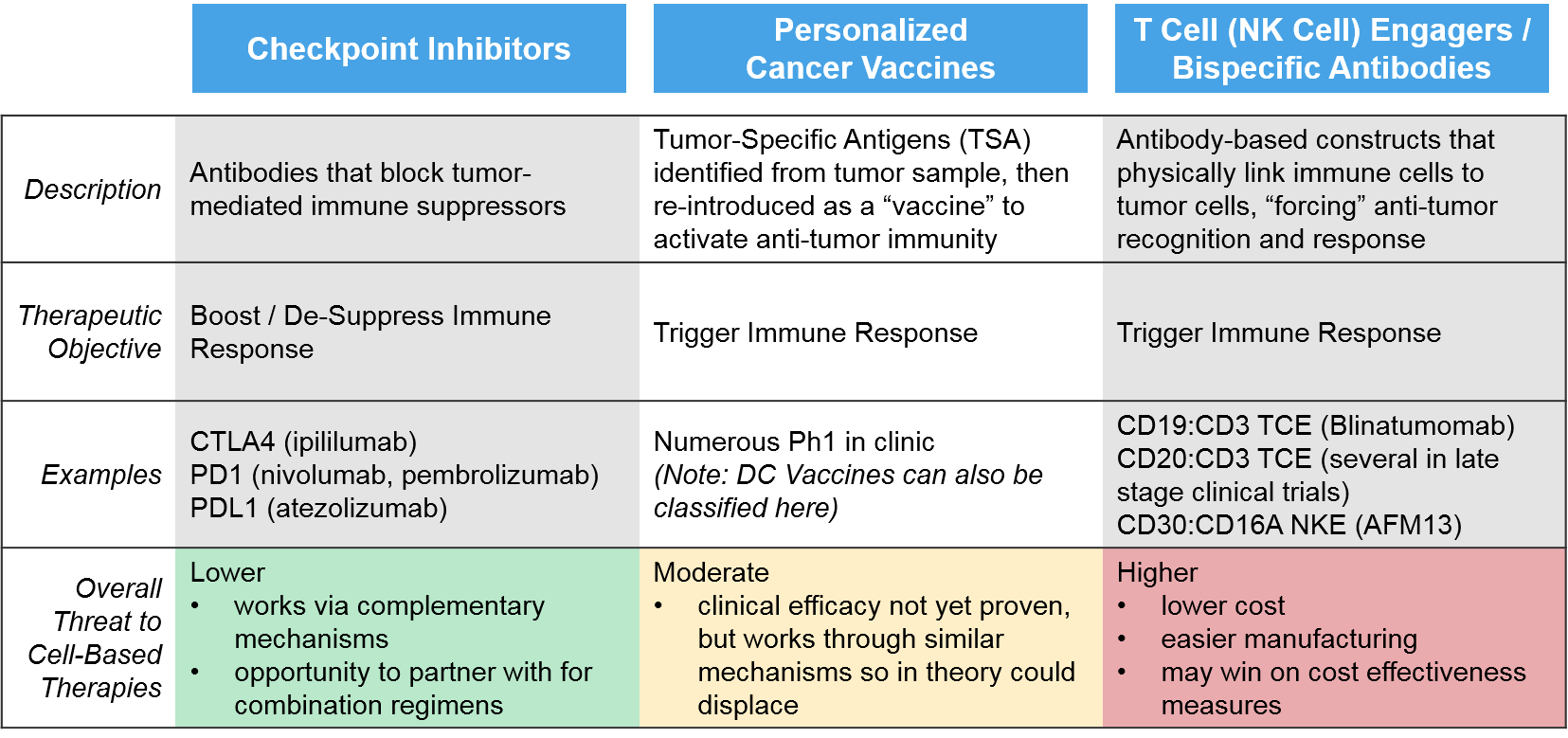
The primary external threats for cell-based therapies are other immunotherapies, since all these products seek to boost and sustain robust anti-tumor immune activation. These include checkpoint inhibitors, personalized cancer vaccines, and T / NK Cell Engagers.
The traditional checkpoint inhibitor immunotherapies work via “unblocking” immune suppression, whereas the newer classes of immunotherapies (T/NK cell engagers, personalized cancer vaccines) work by triggering anti-cancer immunity, as do the cell-based therapies. Because the checkpoint inhibitors work via distinct, complementary mechanisms to the cell-based therapies, they are less of a direct threat, and more of an opportunity as combinatorial or sequential regimen partners. For example, in those immunoreactive tumor types where checkpoint inhibitors are already established, cell-based therapies may be able to enter in later lines (after progression on checkpoint inhibitors) before moving to earlier lines as combination partners.
The personalized cancer vaccines are created by identifying tumor-specific antigens and re-introducing them as a vaccine, either as peptides (protein fragments) or DNA / RNA that encodes those peptides. These products are all still in the clinic with mixed evidence of efficacy. However, if they do ultimately work, they could prove a threat to cell-based therapies, particularly DC Vaccines which work via a very similar mechanism.
The greatest potential threat comes from the T / NK Cell Engagers, which are antibody complexes that target multiple targets at once including an immune cell (T Cell or NK Cell) and a tumor-expressed target (e.g., CD19). This approach is functionally similar to the CAR-T approach, but significantly cheaper and easier to manufacture. A CD19:CD3 T Cell Engager has already been approved (blinatumomab) and several CD20:CD3 T Cell Engagers and a CD30:CD16A NK Cell Engager are in late stage clinical trials.
Summary and Key Takeaways
The cell-based therapy space holds enormous potential for both patients and pharma, but each product must overcome significant challenges to reach approval and maintain a foothold in the marketplace. The successful cell-based therapy developer will design a balanced product that accounts for efficacy, safety, and manufacturing considerations, which on the whole provide a differentiation edge over other cell therapies as well as out-of-class challengers like T /NK Cell Engagers.
Blue Matter Contributors:
- Jeron Eaves
- Pankaj Oza
- Amanda Nottke





