Part One of a Two-Part Series: How Cancer Immunity Works & Types of Cell-Based Immunotherapy
Until recently, cancer treatments for advanced disease were palliative in nature, seeking to reduce tumor burden and prolong life. Curing metastatic disease was not considered possible. But over the past few decades, a new class of cancer treatments has demonstrated curative potential even in late stage patients: cancer immunotherapies.
So far, the approved “checkpoint inhibitor” cancer immunotherapies have been successful in only a subset of tumor types and patients. However, the cancer immunotherapy landscape is expanding to include several new and promising types of immunotherapies, such as cell-based immunotherapies, cancer viruses, and personalized vaccines. In this article (Part One of a Two-part series), we:
- Provide an overview of cancer immunity and how cell-based immunotherapies work
- Review each type of cell-based immunotherapy
In Part Two, we’ll explore the commercial outlook for these therapies. Specifically, we will:
- Place each type into a broader context of therapeutic potential and market competitiveness
- Discuss the development and commercialization outlook for each and the class as a whole
Cancer Immunity: How the Immune System Can Attack Cancer
The immune system’s core function is to differentiate between “self” and “non-self”, and to target and destroy “non-self” entities within the body. These entities can include infectious viruses and bacteria, as well as cancer cells – if they can be recognized as “non-self”.
There are two types of immunity: Innate and Adaptive (historically called Acquired). Innate Immunity is the intrinsic ability of the immune system to mount a rapid response against general classes of targets based on patterns of molecules on the cell surface. The key innate immune cells are called Natural Killer (NK) Cells and Dendritic Cells (DCs), which recognize these patterns and force apoptosis (programmed cell death) upon their targets, then capture the released antigens (protein fragments that the immune system can recognize as either “self” or “non-self”). Innate immunity is fast, but it is fixed on the same limited set of targets; it cannot be triggered for newly-identified “non-self” antigens.
The immune system has a complementary type of immunity, Adaptive Immunity, which refers to the immune system’s ability to identify a previously unrecognized target antigen as “non-self” and mount a highly specific response. The key adaptive immune cells are called Cytotoxic T Lymphocytes (CTLs), or just “T Cells” for our purposes here.
Each immature T Cell has receptors with unique variations that enable it to recognize different potential antigens. No two immature T Cells recognize the same antigen. Adaptive Immunity is prompted when the Innate Immune system presents a novel antigen to an immature T Cell which recognizes and binds to it. This binding triggers activation and amplification of that single T Cell into a population of activated T Cells that roam throughout the body and kill any cell bearing the target antigen. This results in a slower but more targeted response than Innate Immunity, as well as one that “remembers” (if more cells with the target antigen show up in the future, the antigen-specific T Cells will rapidly expand and kill again).
Both types of immunity can play a role in the immune system’s ability to detect and fight cancer. Cancer occurs when the body’s own cells undergo a series of mutations that result in uncontrollable growth and spreading throughout the body. These mutations can change the cancer cell enough that it becomes recognizable as “non-self”, enabling the immune system to detect and destroy tumor cells through Innate and / or Adaptive immune responses. This is the likely cause of at least some cases of reported spontaneous cancer remissions. Understandably, there has been significant interest in how to induce or boost this process via therapeutic interventions.
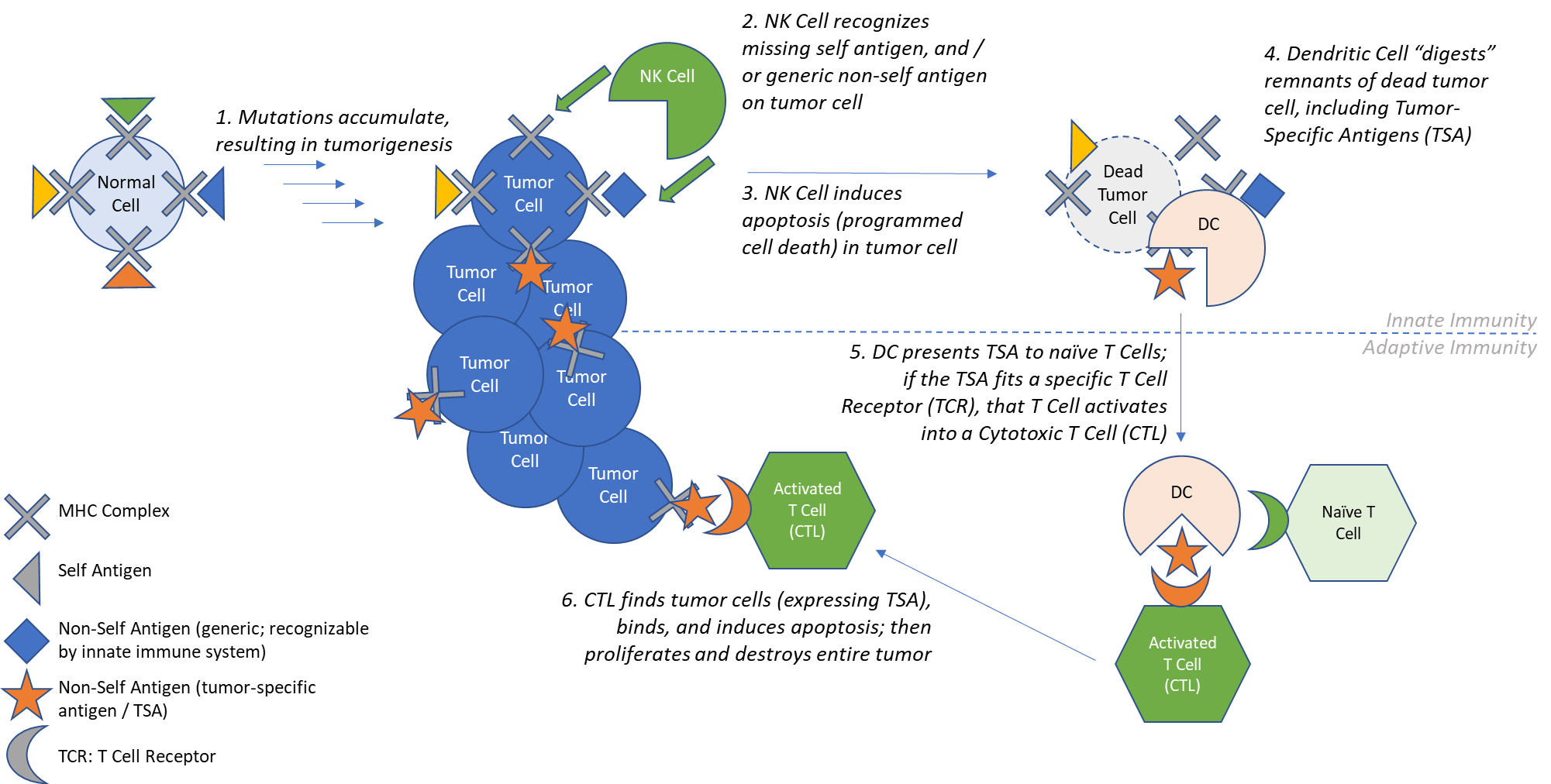
It is very likely that the immune system identifies and destroys many new cancers before they grow and spread. But any cancer that still exists and is being treated is one that has not yet been successfully handled by the immune system. Understanding how cancer immunity fails suggests ways we might assist it therapeutically. The immune system fails to mount a successful defense against cancer in one of two ways:
- No immune response occurs: Cancer cells are not identifiable to the immune system as “non-self” because they do not express surface molecules or present antigens that the innate immune system can recognize, so no adaptive immunity or tumor-specific T Cells are generated.
- Immune response occurs but is evaded: Cancer cells can be initially successfully targeted by the adaptive immune system, but subsequently develop resistance mechanisms. For example, they can establish barriers that block or depress the activity of T Cells or mutate so they no longer present antigens for T Cell recognition (effectively “hiding” from the immune system).
Cancer Immunotherapy: How Therapeutic Interventions Can Trigger and / or Boost Cancer Immunity
Cancer immunotherapy can be loosely defined as any therapeutic intervention which is intended to assist an immune response against the cancer. This includes the historical use of induced infection at tumor sites to attempt spontaneous regression, as well as more recently the checkpoint inhibitor drugs that act by blocking immune suppressor proteins such as CTLA4 and PD1/PDL1. These drugs have been approved in several tumor types including melanoma and lung, with a subset of individuals experiencing deep and durable responses.
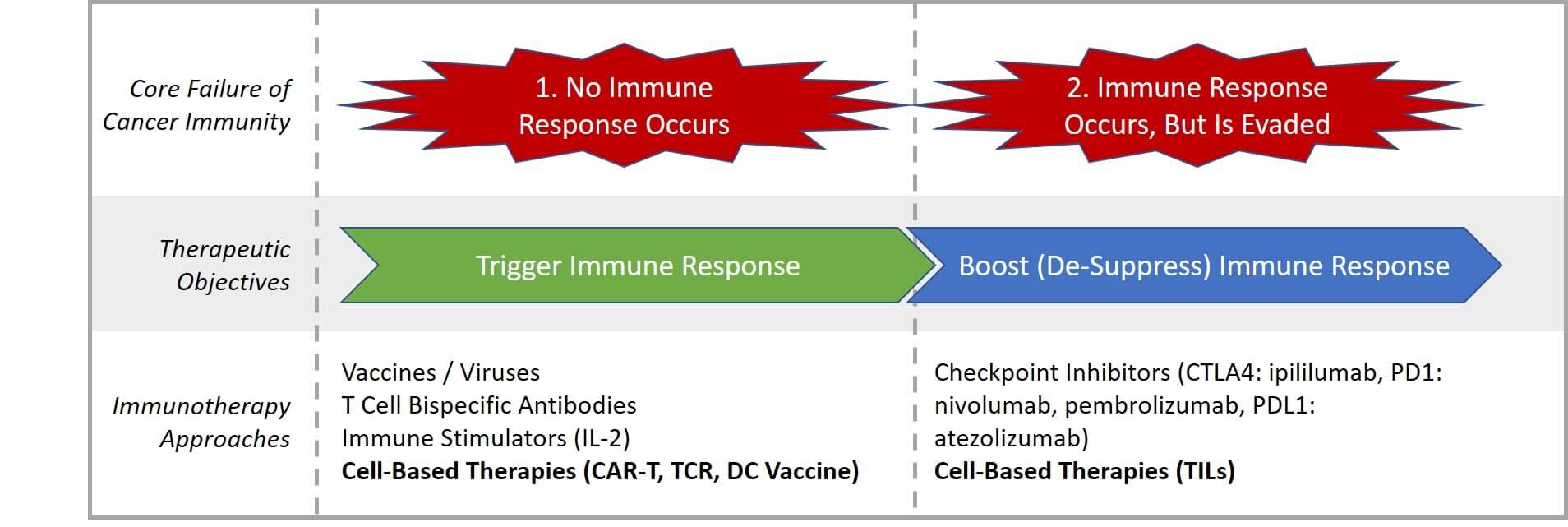
As opposed to classic drug approaches that stimulate or block various molecular pathways, cell-based cancer immunotherapies are actual intact, living immune cells that are removed from the body and either grown to increase their amount and potency, or genetically-modified to boost (or artificially force) their ability to find and kill tumor cells.
The end goal for this approach is to generate and expand tumor-specific T Cells that find, invade, and kill tumors. Cell-based cancer immunotherapies have unique potential because they directly force (or bypass) existing cancer immune pathways to create more effective T Cells, vs working indirectly via molecular pathways as do classic drug-based interventions.
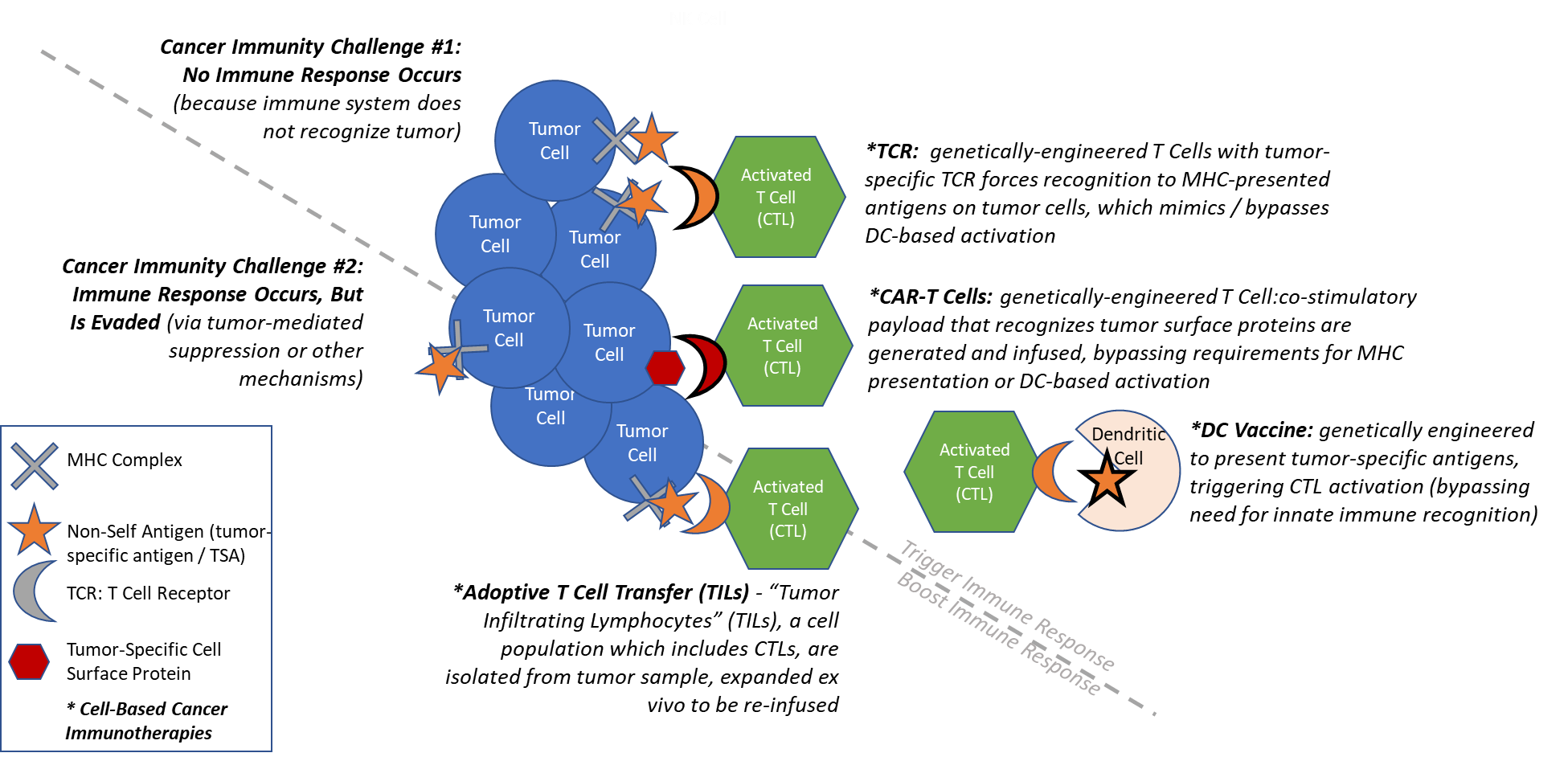
Cell-Based Immunotherapy Classes and Characteristics
Of the numerous cell types playing critical roles in cancer immunity, only two have been extensively targeted for cell-based therapies to-date, namely Cytotoxic T Lymphocytes (T Cells) and Dendritic Cells. This is because these cell types play uniquely direct roles in either classifying a cancer cell as “non-self” to trigger immune response (Dendritic Cells) or targeting and killing a “non-self”-classified cancer cell (T Cells).
In addition to which immune cell type to build from, the other key decision in cell-based immunotherapy design is which tumor antigen to target. There are several important considerations to boost the likelihood of clinical efficacy while minimizing risks of toxicity:
- Efficacy: Antigen must be expressed by the tumor cells as well as recognizable by immune cells to be targeted and killed. Depending on the design of the cell-based immunotherapy, this may mean it is expressed on the cell surface and / or via the classic MHC presentation.
- Safety: Antigen must be differentially expressed by tumor cells but not healthy cells, otherwise serious side effects including death can result as immune cells attack healthy tissue. In practice, this results in three classes of antigens:
- Tissue-Restricted (expressed by tumor cells, only expressed in a small subset of healthy tissues): MAGE, NY-ESO-1, CEA
- Overexpressed (expressed much more in tumor cells than in healthy tissues): HER2, WT1
- Viral (expressed in viral-infected tumor cells only): HPV, EBV
All cell-based immunotherapy approaches work via the same general process of Adoptive Cell Transfer (ACT), with the intent of either boosting or forcing normal cancer immune biology towards a tumor-specific target antigen. ACT involves:
- Isolation: The immune cell type of interest is extracted from the body and isolated, usually from the patient but sometimes from a donor.
- Modification: Immune cells are genetically-engineered to specifically target their actions to tumor cells (note: there are some cell-based immunotherapies that simply isolate and expand existing tumor-targeting immune cells, skipping this step).
- Expansion: Immune cells are stimulated to activate as needed, then grown to large volumes in cell culture.
- Infusion: Immune cells are injected into the patient’s bloodstream, sometimes with supporting immune boosting drugs.
There are four types of cell-based immunotherapies that have been focused on in the clinic to date: CAR-T, TCR, TILs, and DC Vaccines. In addition, there are numerous other types under preclinical or early clinical investigation, e.g., CAR-NK.
CAR-T Cells
 CAR-T Cells are made using living T Cells and are named for their genetically-modified Chimeric Antigen Receptor (CAR) that replaces the normal T Cell Receptor.
CAR-T Cells are made using living T Cells and are named for their genetically-modified Chimeric Antigen Receptor (CAR) that replaces the normal T Cell Receptor.
A CAR is different than the typical T Cell Receptor because it directly binds proteins expressed on the tumor cell surface, whereas the typical T Cell Receptor can only bind antigens that are presented by the MHC proteins. This is an important feature of CAR-T because MHC presentation failure is a common mechanism of tumor resistance to the immune system, which this design circumvents.
CAR-T Cells are made by removing healthy T Cells from either the blood of the patient or a donor, then genetically-modifying them to express a CAR that is specific to a selected tumor cell surface protein (for example, CD19 in leukemias), then infusing them back into the patient’s bloodstream where they circulate throughout the body to find and kill cells expressing the CAR-T target antigen. Because CAR-T Cells can be made from donors, they have the potential for a more “off the shelf” and scalable manufacturing approach than some of the other cell-based immunotherapies.
CAR-T cell design has evolved through several generations to-date. The first-generation CAR-T Cells only expressed the CAR and were not very efficacious because they were not successfully activated once added back into the body. The second generation of CAR-T Cells expressed the CAR, as well as a co-stimulatory molecule designed to prompt immune activation.
This design has been successful in liquid tumors, culminating in two FDA approved products: Novartis’ Tisagenlecleucel “KYMRIAH” (first approved in August 2017), and Kite’s axi-cabtagene ciloleucel “YESCARTA” (first approved in October 2018). Both CAR-Ts target CD19-expressing leukemia cells and include a co-stimulatory molecule to boost efficacy.
CAR-T efficacy has been more mixed in solid tumors, likely due to challenges in appropriate tumor cell surface protein targeting, tumor microenvironment-mediated suppression, and “T Cell exhaustion” (from repeated killing). Because it is difficult to find truly tumor-specific cell surface expressed proteins, there is also an inherent risk of immune attack on healthy tissues that also express whatever marker is targeted.
To address observed and anticipated autoimmune issues, as well as to counter tumor-mediated immune suppression, CAR-T design has expanded into subsequent generations, with third-generation CAR-T expressing multiple co-stimulatory molecules, and fourth-generation CAR-T expressing cytokines (immune boosters) directly. Some CAR-Ts are also being designed to require binding to two distinct tumor antigens to activate (reducing the risks of autoimmune side effects).
CAR-T is currently the most active category of cell-based therapy in clinical development. As of November 2018, about 250 clinical trials are ongoing in both liquid and solid tumors.
TCR
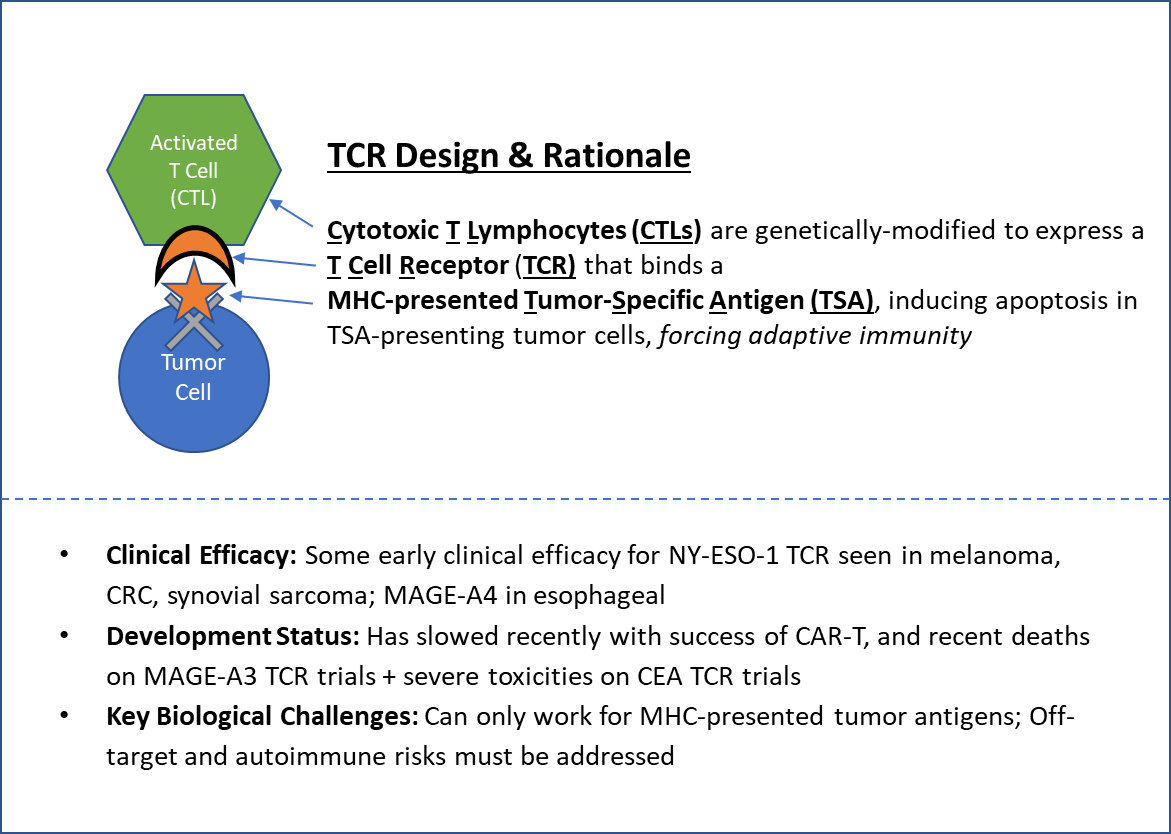 Like CAR-T, T-Cell Receptor (TCR) therapies also involve modified T Cells. The key design difference is that instead of adding in a Chimeric Antigen Receptor, they substitute in a known T Cell Receptor that recognizes a particular MHC-presented Tumor Specific Antigen (TSA).
Like CAR-T, T-Cell Receptor (TCR) therapies also involve modified T Cells. The key design difference is that instead of adding in a Chimeric Antigen Receptor, they substitute in a known T Cell Receptor that recognizes a particular MHC-presented Tumor Specific Antigen (TSA).
Unlike CAR-T, which can theoretically be used somewhat off-the-shelf, TCR cells must be uniquely created for each patient using T Cells from the patient or an MHC-matched donor. Clinical studies have shown some efficacy with a limited set of TSAs first in melanoma, then other solid tumors including synovial sarcoma, as well as esophageal and colorectal cancers.
TCR therapies have a potentially broader applicability than CAR-T because they can be designed to recognize any protein expressed by a tumor cell, not just those that are naturally surface-expressed (as long as it can be presented by the MHC proteins). But while some efficacy has been observed in clinical trials, there have also been severe toxicities and deaths due to immune recognition of healthy tissues. These serious adverse events, as well as the more robust efficacy seen in CAR-T cells, have shifted clinical development focus away from TCR. The requirement for more personalized manufacturing also makes these less appealing from a cost and timing standpoint.
TILs
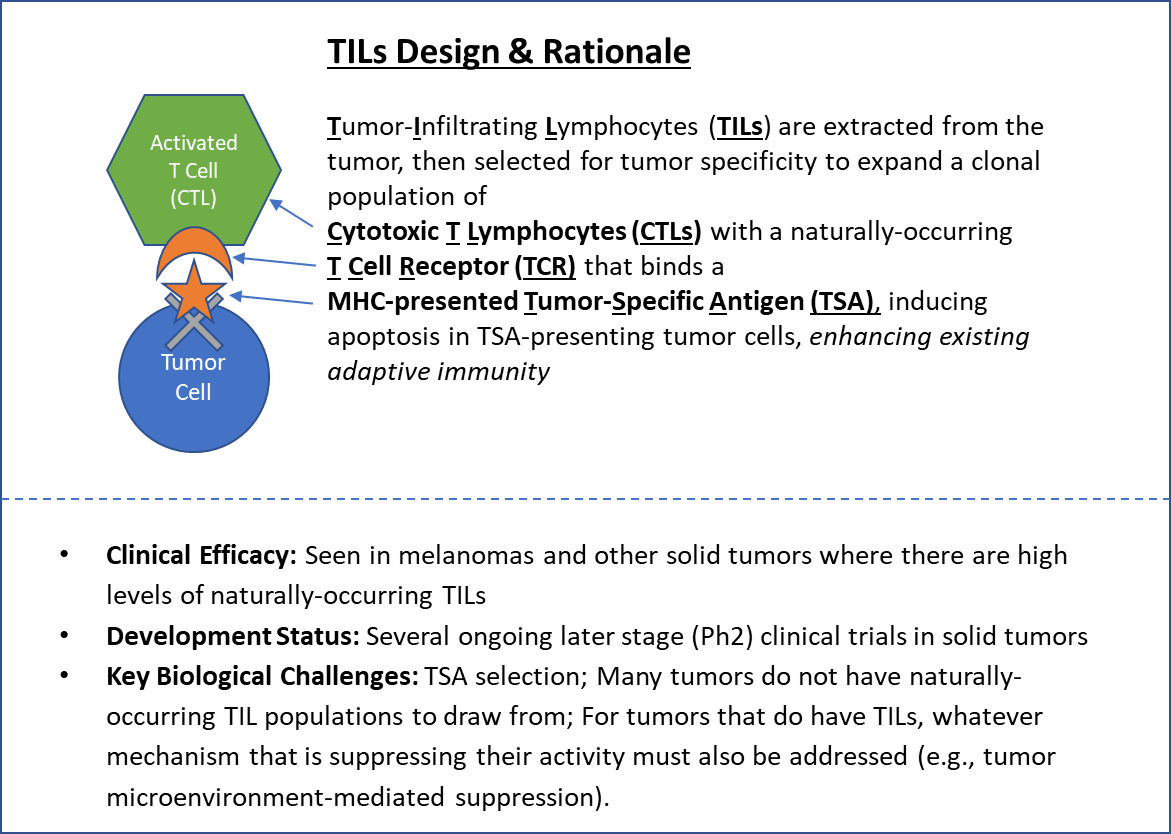 Tumor-Infiltrating Lymphocytes (TILs), a purified population of naturally-occurring T Cells, were the first cell-based therapy successfully attempted. This approach is based on the rationale that simply expanding the number of existing tumor-specific T Cells could be sufficient to overcome resistance to cancer immunity.
Tumor-Infiltrating Lymphocytes (TILs), a purified population of naturally-occurring T Cells, were the first cell-based therapy successfully attempted. This approach is based on the rationale that simply expanding the number of existing tumor-specific T Cells could be sufficient to overcome resistance to cancer immunity.
Immune cells are extracted from the inside the tumor, then selected against either known Tumor Specific Antigens (TSA) or against cell lines generated from the tumor itself. The tumor-specific T Cells purified via this screening are then expanded via cell culture and reinfused into the patient along with the immune booster IL-12.
TILs infusion can be effective for some patients with melanomas, as well as a few other tumor types including breast. Several later stage (Ph2) clinical trials are ongoing.
Because TILs are obtained from the patient, they are typically very specific and targeted to the tumor, so the risk of adverse events from inappropriate immune responses is hypothetically lower. However, not all tumor types or individual patients have a suitable population of TILs to draw from, which has limited the scope of this therapy.
DC Vaccines
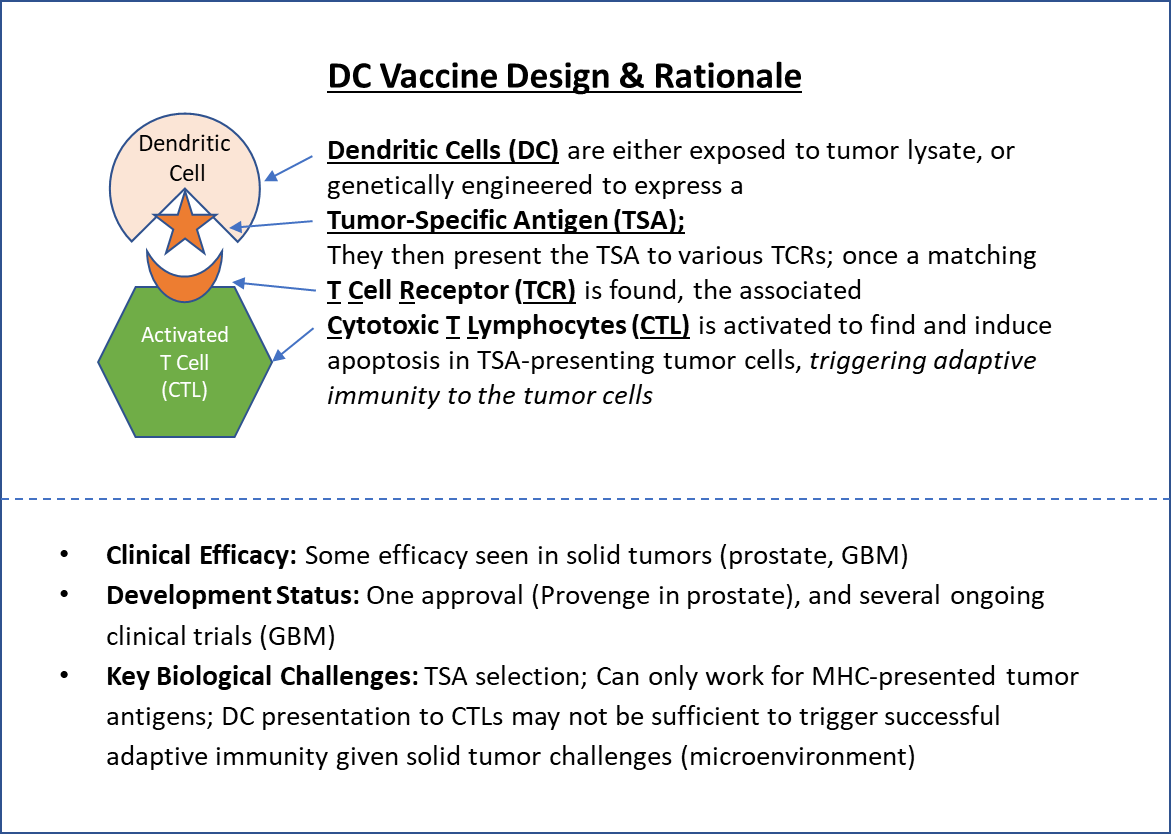 Unlike TILs, TCR, and CAR-T Cells, DC vaccines do not involve direct manipulation of T Cells. Instead, the focus is modulating the adaptive immunity trigger of Dendritic Cell-mediated antigen presentation. To create a DC Vaccine, Dendritic Cells are extracted and either “primed” to Tumor-Specific Antigens (TSA) by exposing them to tumor cell lysate, or genetically-engineered to directly express a chosen TSA. Then, once they are added back into the body, they present those TSAs to naïve T Lymphocytes, triggering the adaptive immune response. This is similar to how vaccines work, hence the name.
Unlike TILs, TCR, and CAR-T Cells, DC vaccines do not involve direct manipulation of T Cells. Instead, the focus is modulating the adaptive immunity trigger of Dendritic Cell-mediated antigen presentation. To create a DC Vaccine, Dendritic Cells are extracted and either “primed” to Tumor-Specific Antigens (TSA) by exposing them to tumor cell lysate, or genetically-engineered to directly express a chosen TSA. Then, once they are added back into the body, they present those TSAs to naïve T Lymphocytes, triggering the adaptive immune response. This is similar to how vaccines work, hence the name.
One DC therapy has been FDA-approved (Dendreon’s sipoleucel-T “PROVENGE” in prostate cancer), but like other cell-based immunotherapy approaches, efficacy has been mixed in other solid tumors. There has also been controversy about the high price given the relatively modest efficacy ($100,000 for a 4-month increase in median overall survival), which also suggests that DC vaccines may not share the curative potential of other cell-based immunotherapies. Several clinical trials are ongoing for other DC Vaccines, including some in glioblastoma, that are reporting promising early signs of durable efficacy.
Looking Ahead
Hopefully, this article has provided a helpful overview of the different types of cell-based therapies that are currently being developed and used to fight cancer. In Part Two, we’ll take a closer look at the commercial opportunities and challenges these therapies face.





